参考来源1:https://www.jianshu.com/p/5d7e550abc1a
参考来源2:https://www.jianshu.com/p/971ff2ee533a
参考来源3:https://www.plob.org/article/10843.html
第一部分 DNA甲基化
1. DNA甲基化背景知识
DNA甲基化是表观遗传学(Epigenetics)的重要组成部分,在维持正常细胞功能、遗传印记、胚胎发育以及人类肿瘤发生中起着重要作用,是目前新的研究热点之一。DNA甲基化(DNAmethylation)在基因表达调控方面发挥重要作用,并与转座子和其他重复原件的沉默有关。异常的DNA甲基化和包括癌症在内的多种疾病有关。
DNA甲基化是发生在DNA序列上的化学修饰——在DNA甲基转移酶(DNMT)的催化下,胞嘧啶的第5个碳原子被添加一个甲基形成5mC。5mC可以被Tet蛋白进一步氧化成5hmC(羟甲基化)。通常所说的DNA甲基化是指的5mC和5hmC。植物体内胞嘧啶的甲基化则可分为对称的CpG(或CpNpG),或是不对称的CpNpNp形式(C与G是碱基;p是磷酸根;N指的是任意的核苷酸)。
特定胞嘧碇受甲基化的情形,可利用亚硫酸盐测序(bisulfite sequencing)方式测定。DNA甲基化可能使基因沉默化,进而使其失去功能。此外,也有一些生物体内不存在DNA甲基化作用。
2. DNA甲基化测序原理及方法(简单介绍)
DNA甲基化测序方法按原理可以分成三大类:
-
1)重亚硫酸盐测序;
-
2)基于限制性内切酶的测序;
-
3)靶向富集甲基化位点测序;
基于以上原理,目前有10种常用的DNA甲基化测序方法:
- 1)重亚硫酸盐测序
- 2)重亚硫酸盐处理后接头标记技术(PBAT)
- 3)限制性内切酶-重亚硫酸盐靶向测序(RRBS)
- 4)氧化-重亚硫酸盐测序(oxBS-Seq)
- 5)TET辅助的重亚硫酸盐测序(TAB-seq)
- 6)甲基化敏感性的限制酶测序(MRE-Seq)
- 7)HELP-Seq
- 8)甲基化DNA免疫共沉淀测序(MeDIP)
- 9)甲基化结合域捕获技术(MBD-CAP)
- 10)基于探针的靶向富集技术
总结来说,目前常用的检测DNA甲基化的技术一般有MeDIP-Seq,MBD-Seq,BS-Seq等,其中BS-Seq可以在单碱基水平上检测DNA甲基化状态,而其他几个只能测得某个区域的甲基化状态。DNA经过亚硫酸氢钠处理后会把非甲基化的胞嘧啶转化为尿嘧啶,但是甲基化的胞嘧啶没有变化。所以经过PCR扩增之后非甲基化的胞嘧啶变为胸腺嘧啶,甲基化的胞嘧啶保持不变。WGBS数据是精确到每个位点的深度测序,数据量因此也很大,动辄就上百个G。
3. 数据分析流程(简单介绍)
3.1 BS-seq分析总览
这里选取2012发表在Nature Methods杂志是一篇综述文章:DNA methylome analysis using short bisulfite sequencing data 总结的数据分析流程,如下:
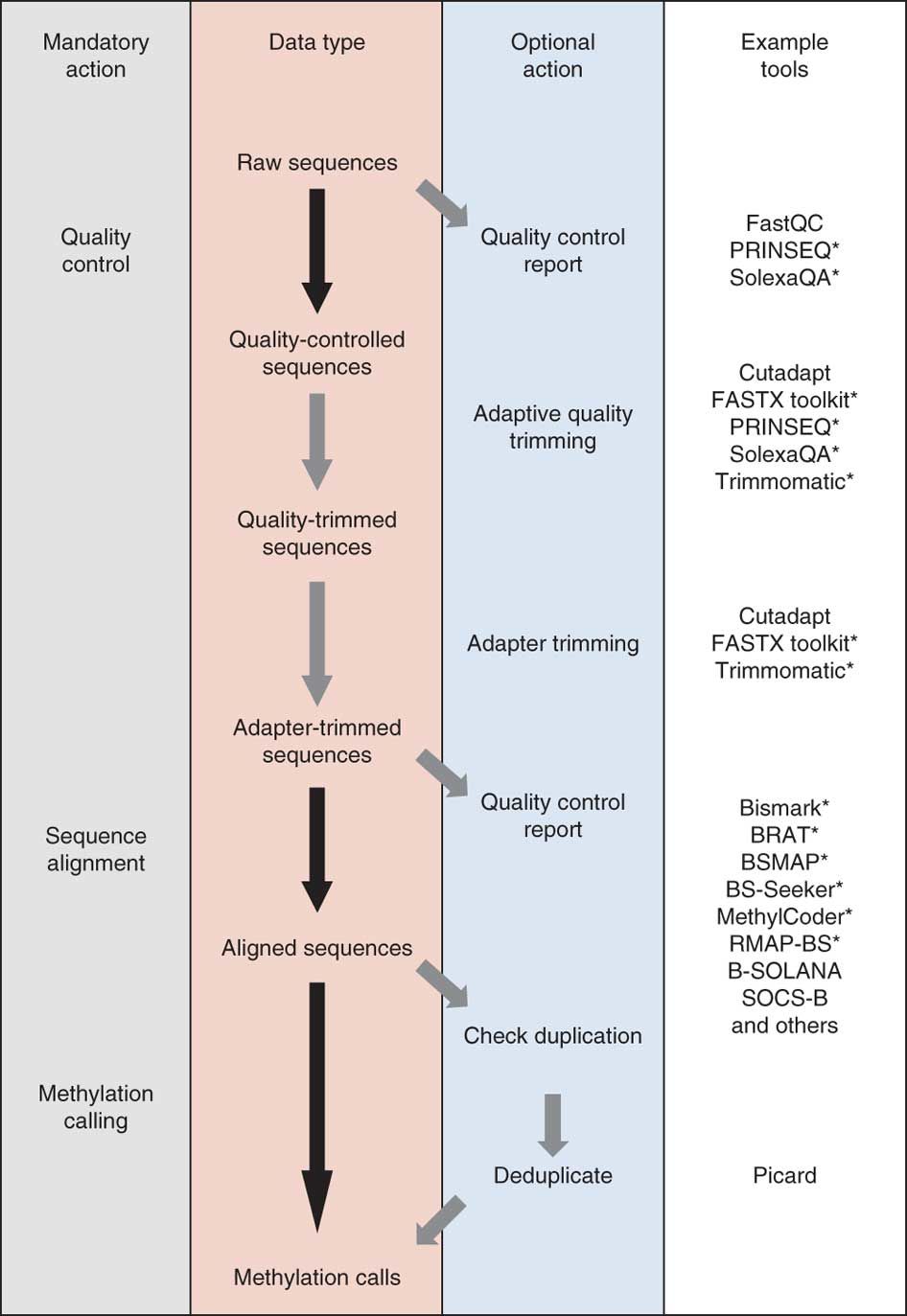
3.2 示例数据来源
这里随便挑选UCSD Human Reference Epigenome Mapping Project中的一个样本GSM429321,是Whole genome shotgun bisulfite sequencing of the H1 cell line,包含了Runs: 43 runs, 570.4M spots, 49.6G bases, 32.3Gb。
主要还是下载数据,数据质量控制,比对,只不过这个流程的比对工具比较特殊,这里使用bismark工具,该工具需要构建参考基因组,然后才能将数据比对到基因组上,本例中使用hg19参考基因。
参考基因组下载地址:http://hgdownload.soe.ucsc.edu/goldenPath/hg19/chromosomes/
3.3 bismark原理
- 使用bismark构建参考基因组:
bismark_genome_preparation --path_to_bowtie /bowtie2-2.0.5 -verbose out/
out/ 为chr19.fa.gz所在目录并且输出结果到该目录。
-verbose 输出更详细的结果和错误调试信息。
--path_to_bowtie bowtie的安装目录,如果不加该参数,稍后会有提示。
- 用bismark将reads比对到构建的参考基因组上:
bismark Y/ --path_to_bowtie …/bowtie2-2.0.5--bowtie2 test.fastq -o align_result
--bowtie2/ --bowtie1 为--bowtie2/--bowtie1建立亚硫酸盐索引
# test.fastq 要比对到参考基因组上的reads文件。
# -o 在align_result文件夹中输出结果文件test_bismark_bt2.bam。
- 根据比对结果test_bismark_bt2.bam计算甲基化水平
bismark_methylation_extractor-s -comprehensive --bedGraph --counts align_result/test_bismark_bt2.bam -o output_file/
# -s/--single-end 指定数据为单端测序
# -p/--paired-end 指定数据为双端测序
# --comprehensive 添加该参数将把4个可能的链特异甲基化信息加入输出文件中。
# --bedGraph –counts 生成bedGraph文件,可以用来得到全基因组范围的胞嘧啶报告。