来源:Illumina官网提供的《单细胞测序工作流程》。
引言
活组织由多种类型的细胞组成,每种细胞类型都有不同的谱系和独特的功能,对组织和器官生物学产生影响, 并最终定义机体整体的生物学功能。每个细胞的谱系和发育阶段决定细胞如何对其他细胞和微环境产生应答。此外,由于随时间产生的随机变化,相同类型细胞的亚群之间以及它们与其他细胞类型之间通常具有遗传异质性。由于存在这种复杂性,想要通过分析大量组织或细胞来深入了解细胞功能非常困难,这就突显出分离单细胞进行研究的必要性。
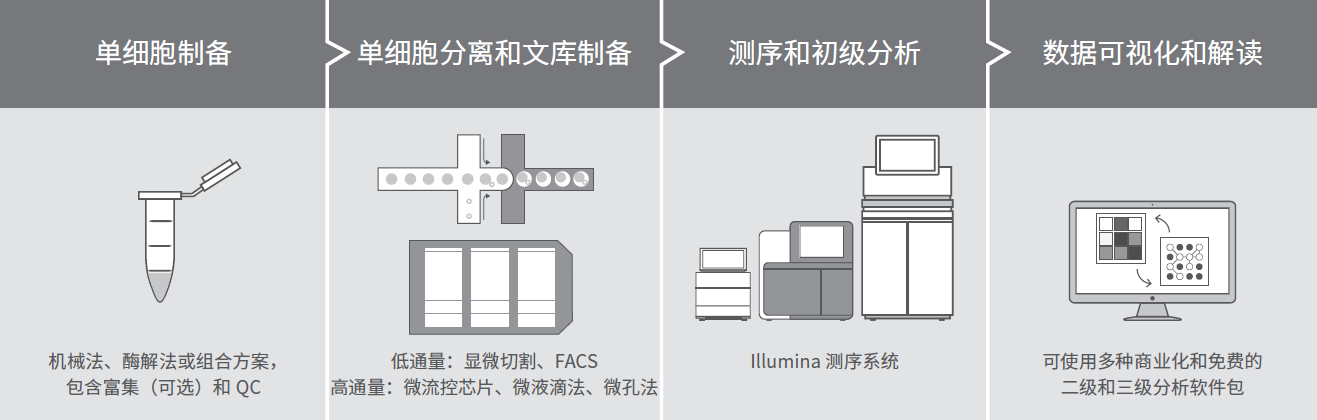
单细胞测序的工作流程:
- 1)单细胞制备
- 2)单细胞分离和文库制备
- 3)测序和初级分析
- 4)数据可视化与解读
第1步:单细胞制备
1.1 裂解

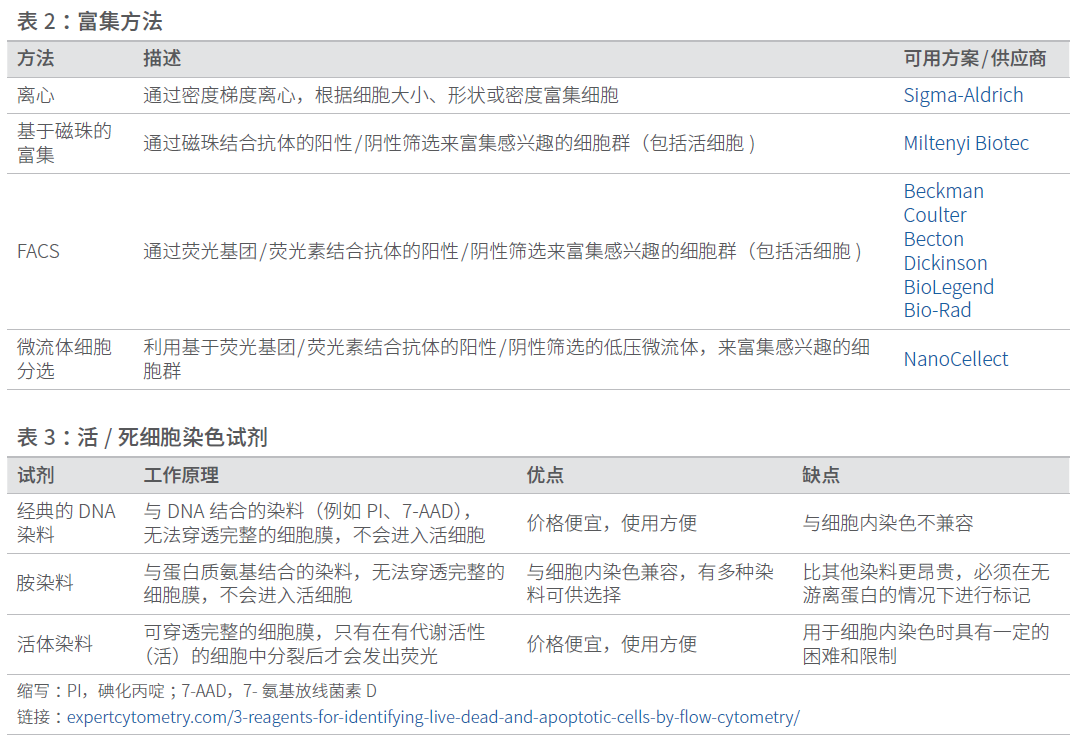
1.2 富集
1.3 质量控制
单细胞测序实验需要投入大量的时间、金钱、样本材料和资源。在进行单细胞分离、文库制备和测序之前,几个简单的质量控制(QC)措施可以确保实验的高质量。
- 目视检查

-
流式细胞术
-
关键指标

第2步:单细胞分离和文库制备
2.1 单细胞分离方法和平台
单细胞分离方法可以根据通量来区分。低通量方法包括手工操作或细胞分选/分离技术(例如FACS),每次实验能处理几十个,几百个甚至几千个细胞(表 4)。微流体技术的进展使高通量单细胞分析成为可能,利用该技术,研究人员可以在一次实验中经济有效地检测成千上万个细胞(表 5)。
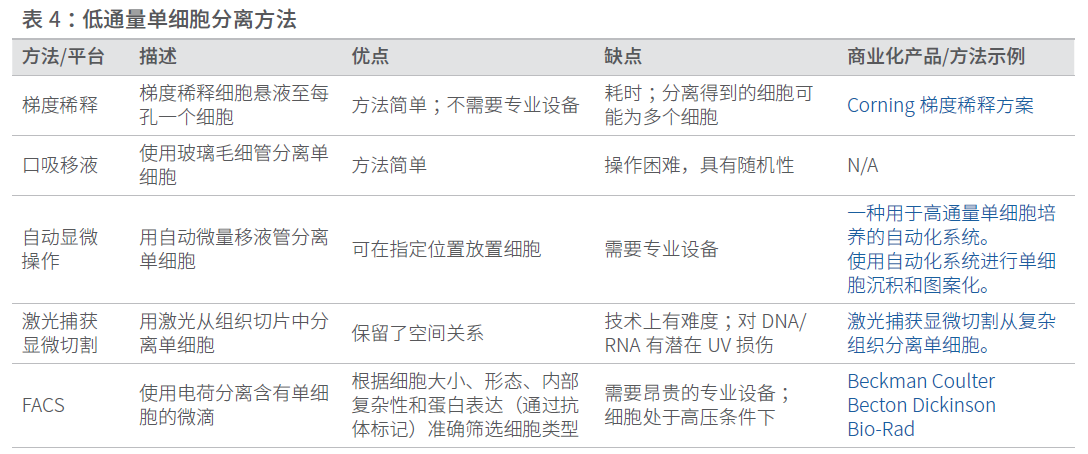
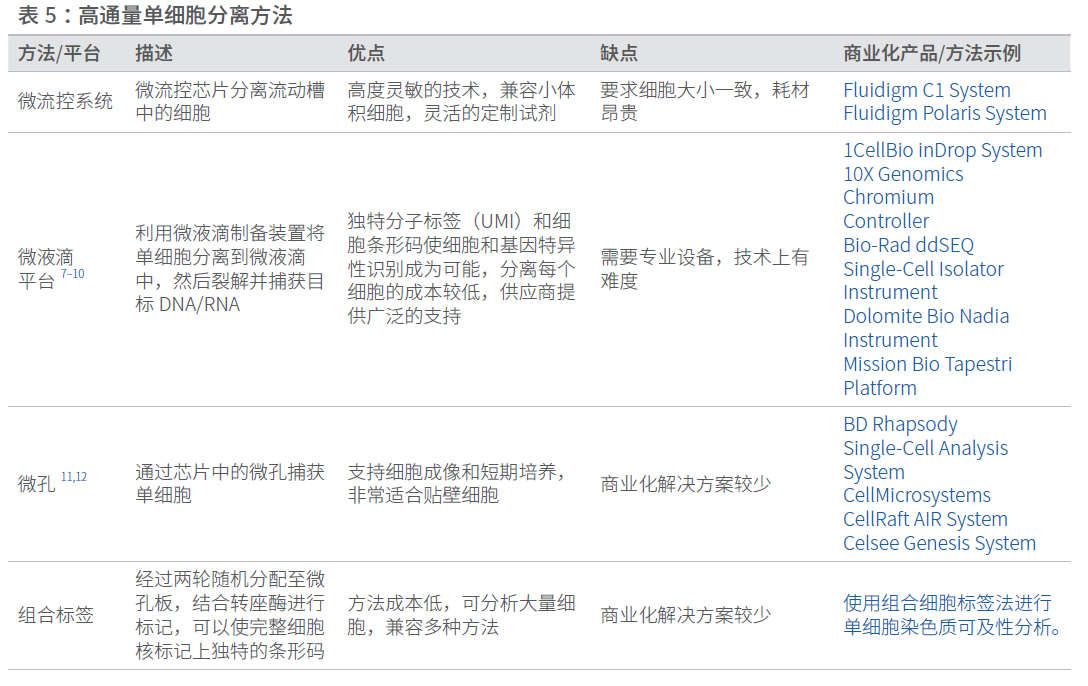
2.2 文库制备
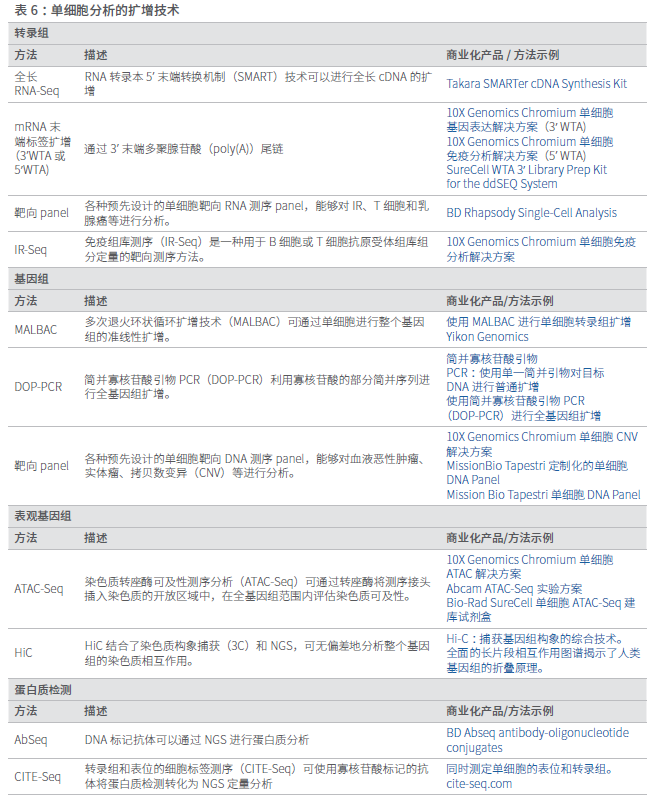
单细胞测序工作流程的下一个关键步骤是文库制备。有多种可用的方法,但选择的细胞分析方法和特定的测序方法是重要的考虑因素(下表)。具体方法的选择在很大程度上将由研究的问题决定。
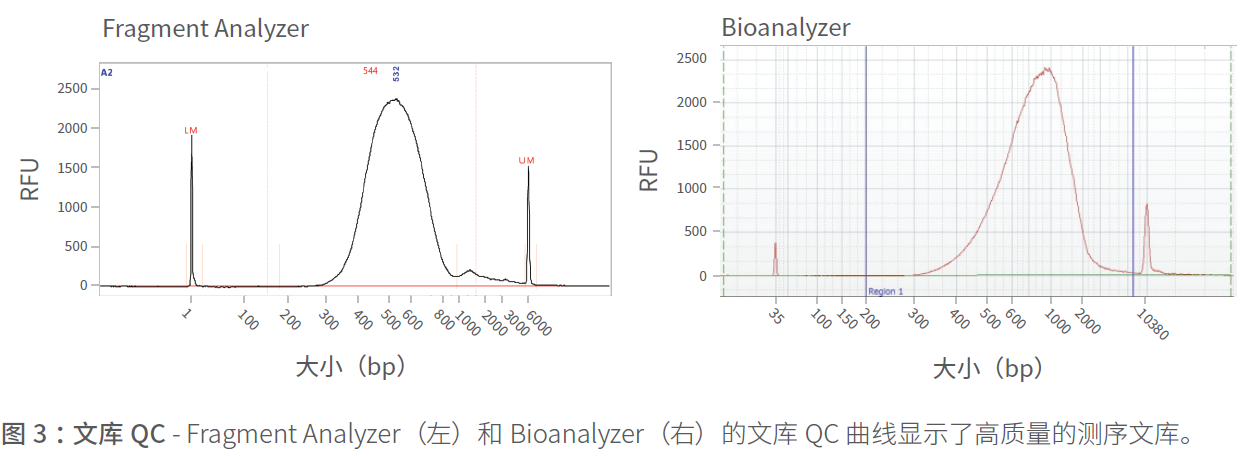
2.3 制备文库的QC
第3步:测序
为了平衡成本和能够达到实验目标的最佳测序参数,应在进行测序之前考虑所需的read 深度,无论单端测序还是双端测序都需要考虑,这一点非常重要。在测序之前,还应该考虑其他测序指标,例如簇密度、%PF 和%≥Q30(以及替代指标),测序后也应评估这些指标,以帮助确保获得成功的结果。获得高质量、可靠的测序数据后,研究者即可进行数据分析、可视化和解读。
第4步:数据分析、可视化和解读
单细胞测序实验的分析流程包括三个阶段:初级分析(碱基检出)、二级分析(多重分离、比对和遗传鉴定)和三级分析(数据可视化和解读)。
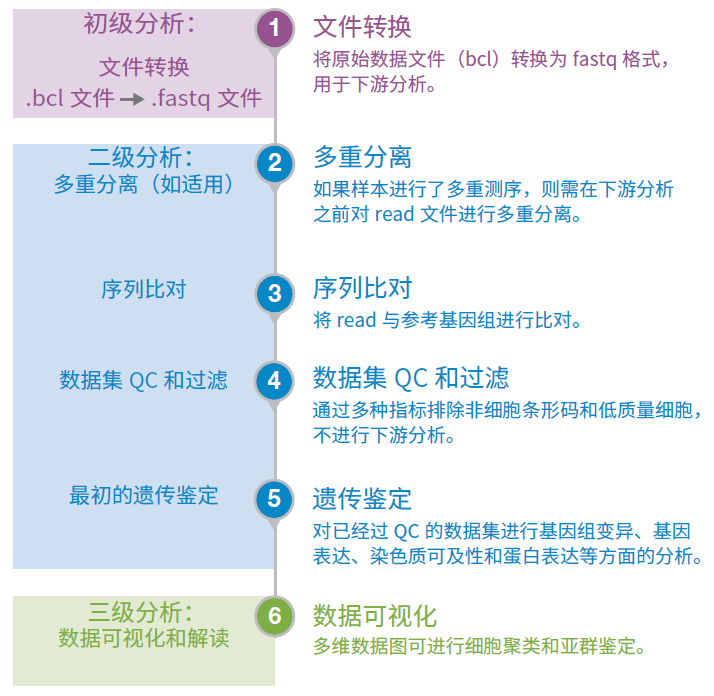
4.1 初级分析:文件转换
*.bcl格式
Illumina 测序系统生成的原始数据文件为二进制碱基检出(BCL)格式。这种测序文件格式包含碱基检出和每个循环中每个簇的碱基检出质量。BCL 文件格式是在测序系统中使用的文件格式,需要转换为FASTQ 格式,才能用于用户开发的或第三方数据分析工具。
*.fastq文件格式
FASTQ 是基于文本的测序数据文件格式,可储存原始测序数据和质量分值。FASTQ 文件已成为储存Illumina 测序系统的NGS 数据的标准格式,可用作多种二级数据分析解决方案的输入。
bcl2fastq转换软件
bcl2fastq 软件能将BCL 文件转换为FASTQ 文件用于下游分析,第一个read 完成测序后即可开始这个过程。如果需要对样本进行多重分析,那么生成FASTQ 文件的第一步就是多重分离。多重测序使多个单独的样本可以在一个流动槽的一个通道中运行,极大地提高了系统的产出。多重分离可以根据簇的标签序列,将簇分配到一个样本。多重分离完成后会根据每个样本组装好的序列生成FASTQ文件。如果样本不进行多重分析,则不用进行多重分离步骤,每个流动槽通道中的所有簇都会分配到一个样本。
4.2 二级分析:多重分离、比对和QC
数据分析的第一步通常是将read 定位和比对到参考基因组中。有多种软件应用可以使用,包括BWAAligner BaseSpace App 使用的Burrows-Wheeler 比对(BWA)算法, 以及RNA-Seq AlignmentBaseSpace App 使用的剪接转录本比对至参考基因组(STAR)算法。
4.3 数据分析QC指标
在进行下游分析之前,应评估多个QC 指标,帮助确定单细胞测序数据集的质量,过滤掉质量较低的数据点/细胞。
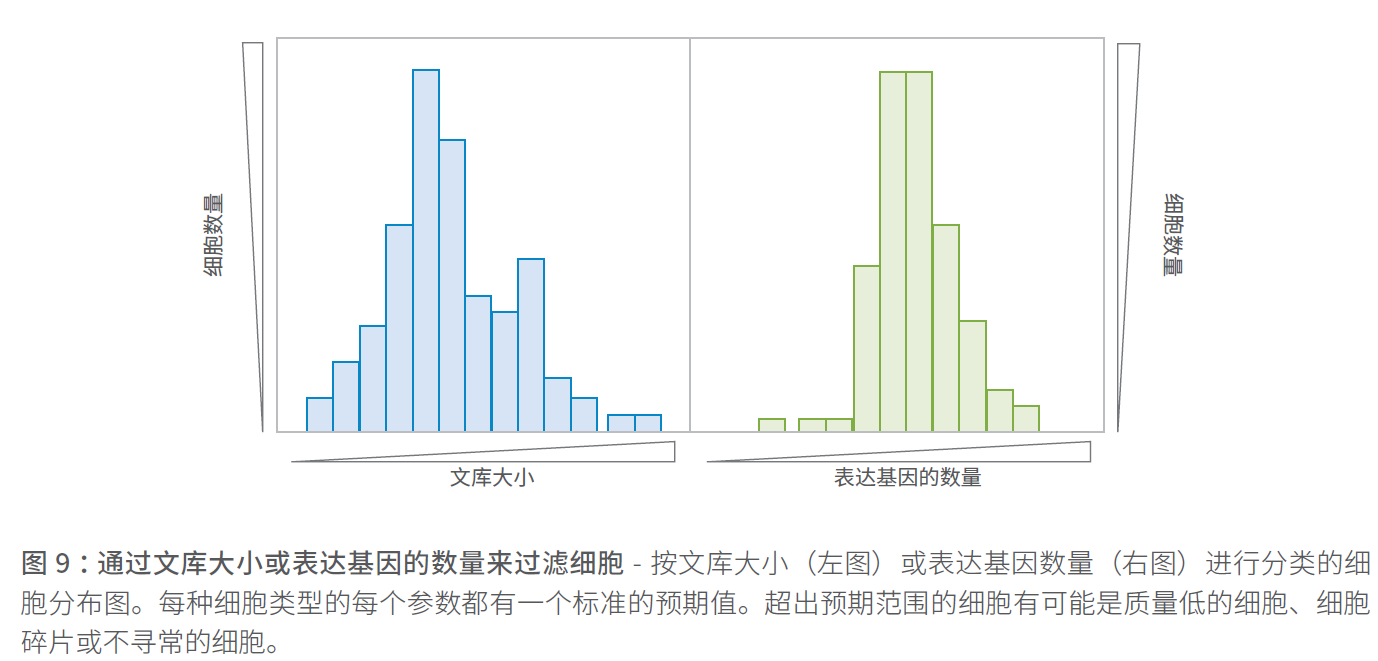
####预期文库大小和表达基因的数量
每种细胞类型都有一个预期的文库大小,对于RNA-Seq,也有一个标准的表达基因数量。位于标准的预期范围之外的细胞(过低或过高)可能代表无需进行下游分析的低质量“细胞”,或者也有可能代表值得进一步研究的不寻常的细胞(下图)。
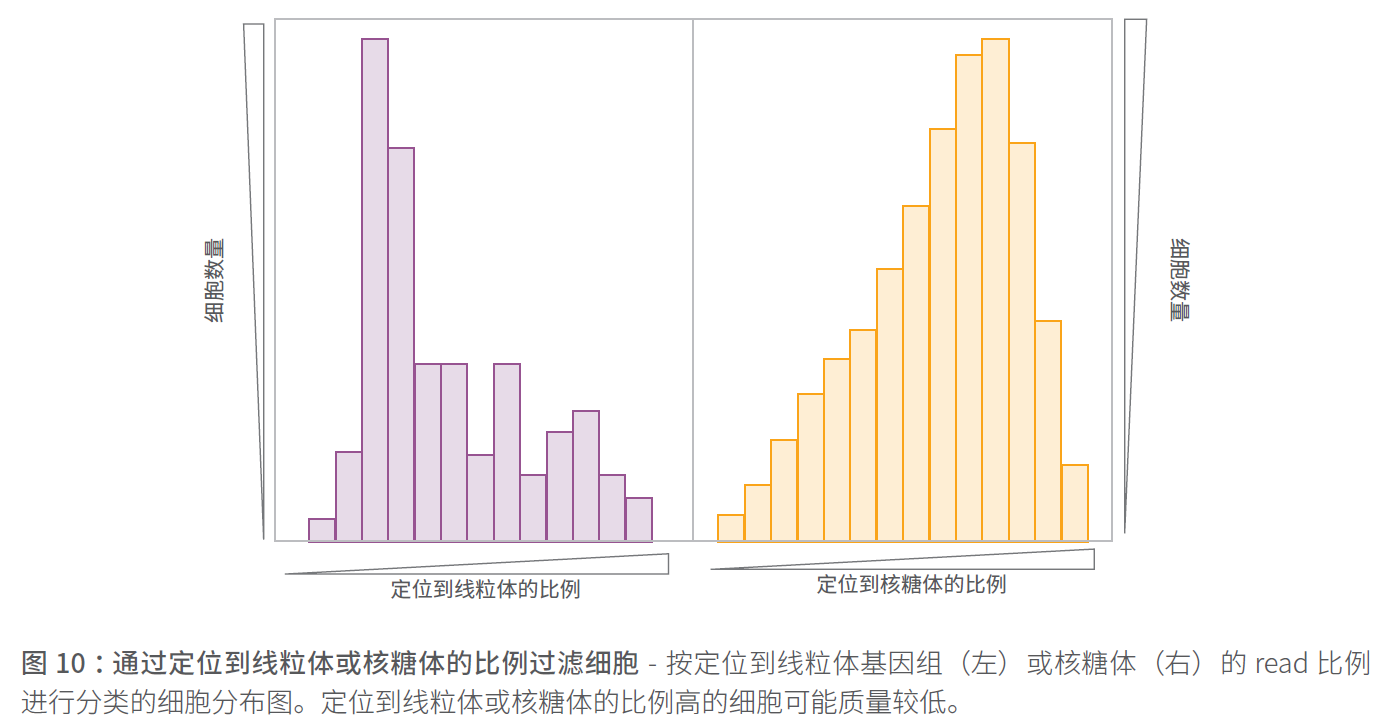
比对到线粒体/核糖体的read比例
另一个QC 指标是定位到线粒体基因组基因的read 比例,或者定位到核糖体RNA 的read 比例(下图)。定位到线粒体和核糖体的比例高表明细胞质量较低,这可能是细胞凋亡增加所致,可以不进行下游分析。
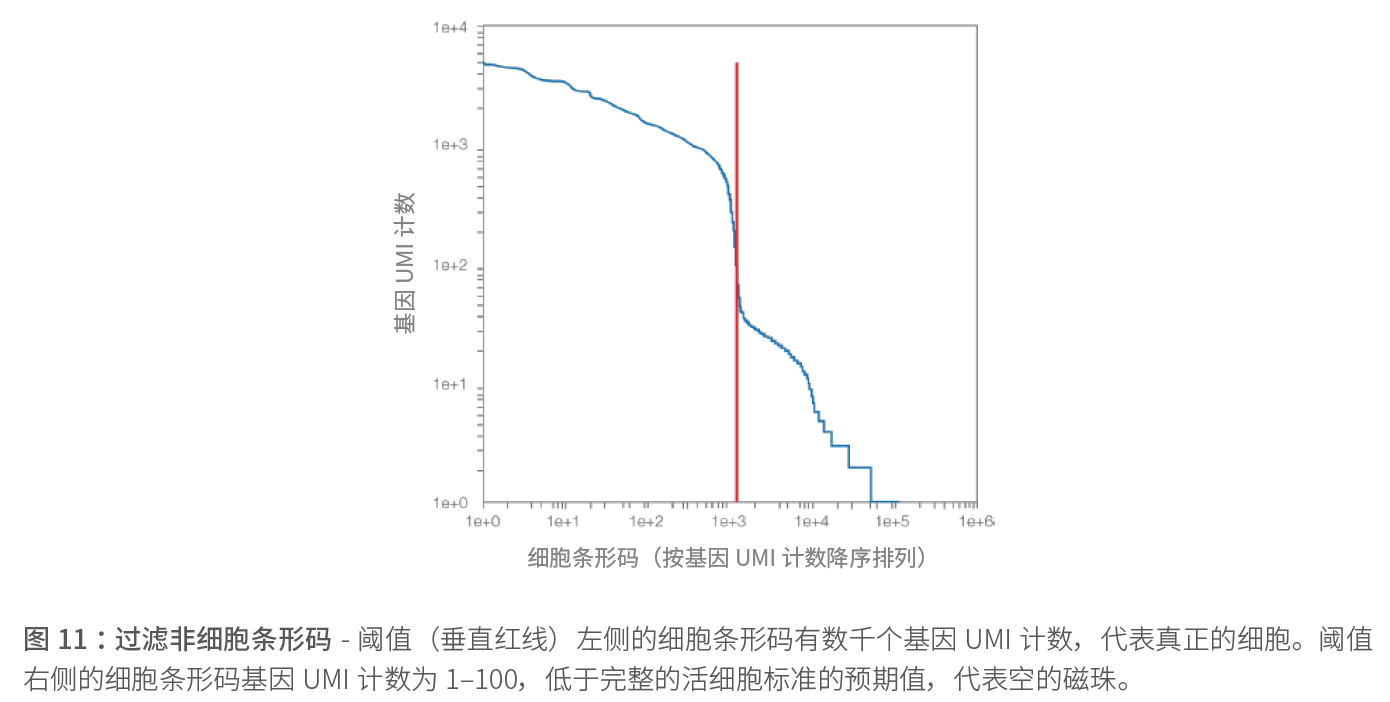
Knee曲线图
绘制基因UMI 计数与细胞条形码(按基因UMI 计数降序排列)的曲线图,可以从统计水平上识别“真正”的细胞,排除非细胞条形码(下图)。阈值以上的细胞条形码(knee 曲线图左侧)的基因UMI 计数代表了真正的细胞,而阈值以下(knee 曲线图右侧)的细胞基因UMI 计数低于特定细胞的预期值。
4.4 评估双联体
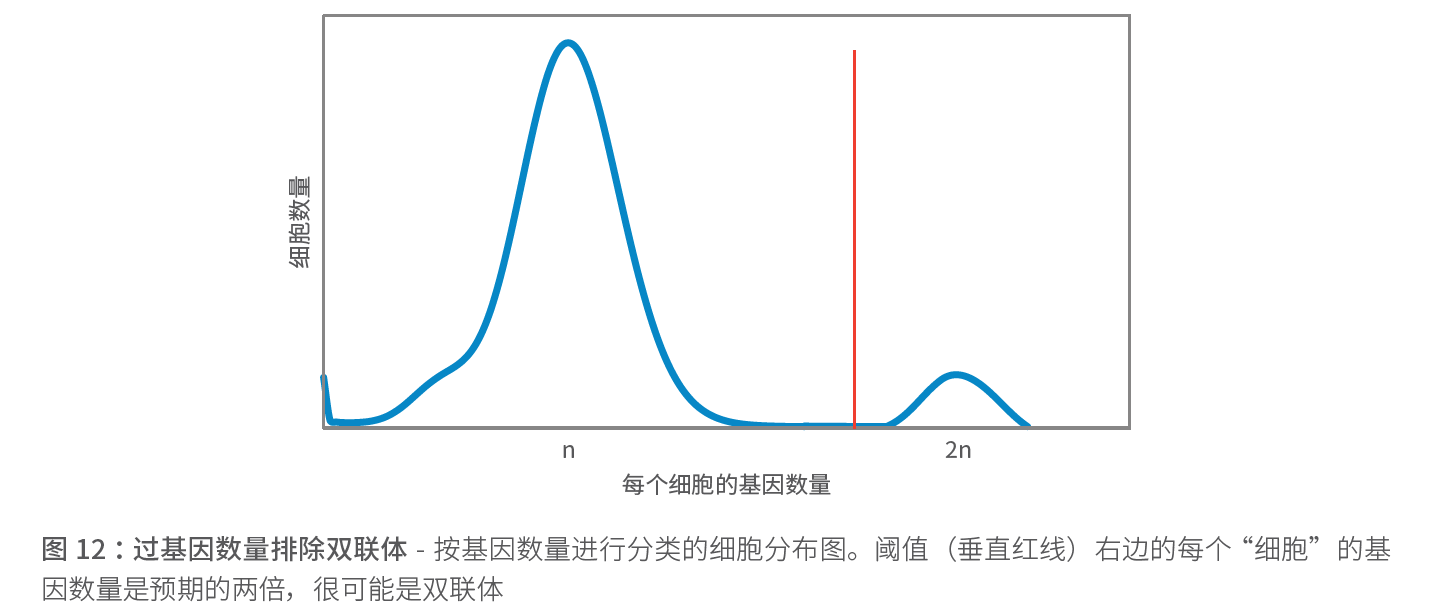
每个细胞的基因数量
任何给定的细胞类型都有一个标准的预期表达基因数量。这个指标已被用于在下游分析中检测和排除双联体。然而,尽管每个细胞使用的基因数可以用于具有同质细胞群的单细胞测序实验,例如培养的细胞系,但它用于复杂的异质组织时可能会有问题。事实上,大多数活的单细胞会落在预期的表达基因数量范围之内(n),分布在范围之外的细胞(例如大约是该数量的两倍,2n)有可能代表感兴趣的细胞,值得进一步研究和鉴定,例如血液样本中的循环肿瘤细胞(下图)。最后,由于缺乏可靠的双联体检测计算方法,研究人员应该通过实验设计最大限度地降低双联体出现的概率。
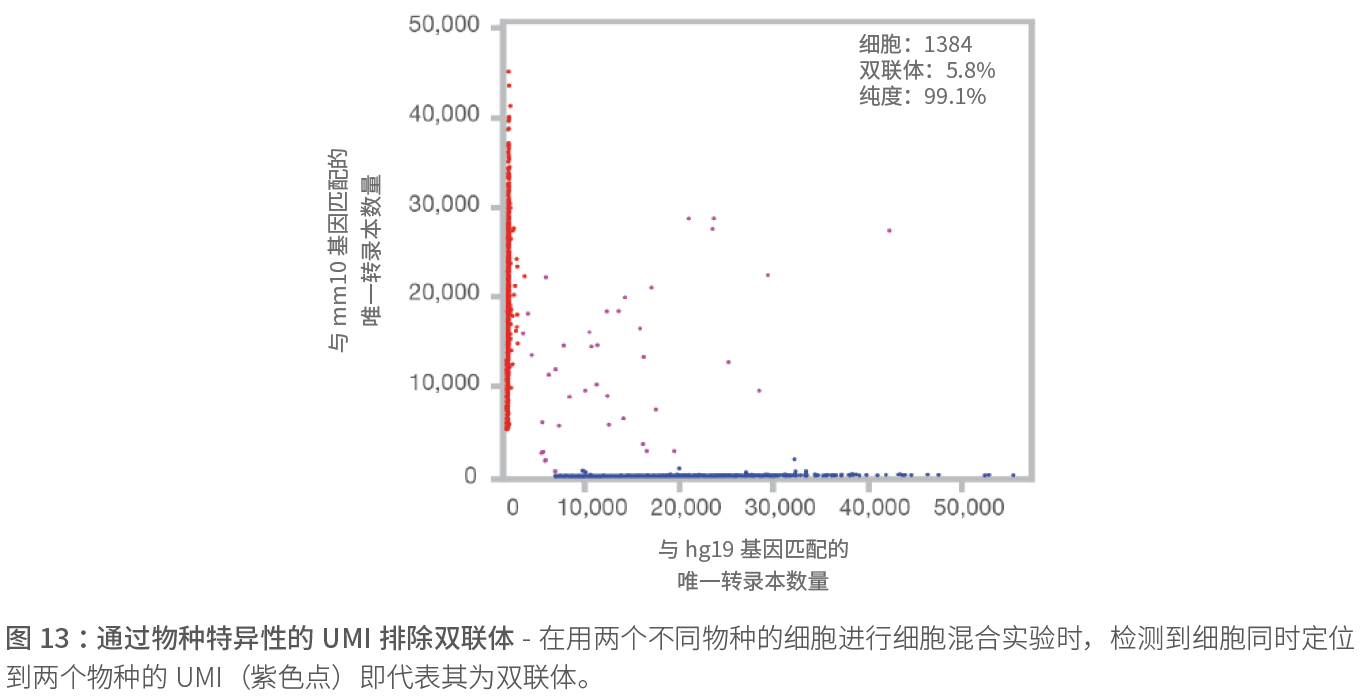
跨物种分析
串扰代表给定实验中液滴或微孔内为双联体细胞的百分比。将两个不同物种的细胞按1:1 的比例混合在一个样本中是一种检测细胞串扰的有效方法。如果分析的样本中包含了两个不同物种的细胞,检测到一个细胞具有两个物种的UMI 时,即代表该细胞为双联体。
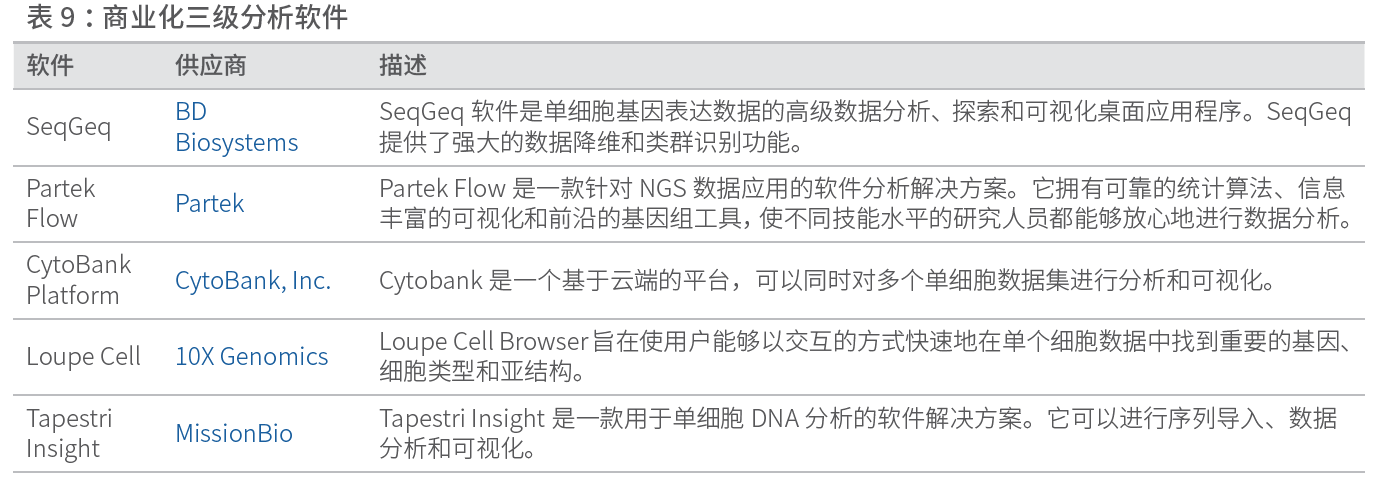
4.5 三级分析:数据可视化和解读
read 比对到参考基因组,并且完成了二级分析(包括数据质量控制以去除非细胞条形码和/ 或质量较低的细胞)后,就可以对质量较好数据集进行可视化和深入探索,从而深入了解所研究细胞的生物学信息。有许多免费的和商业化的软件程序可以使用。
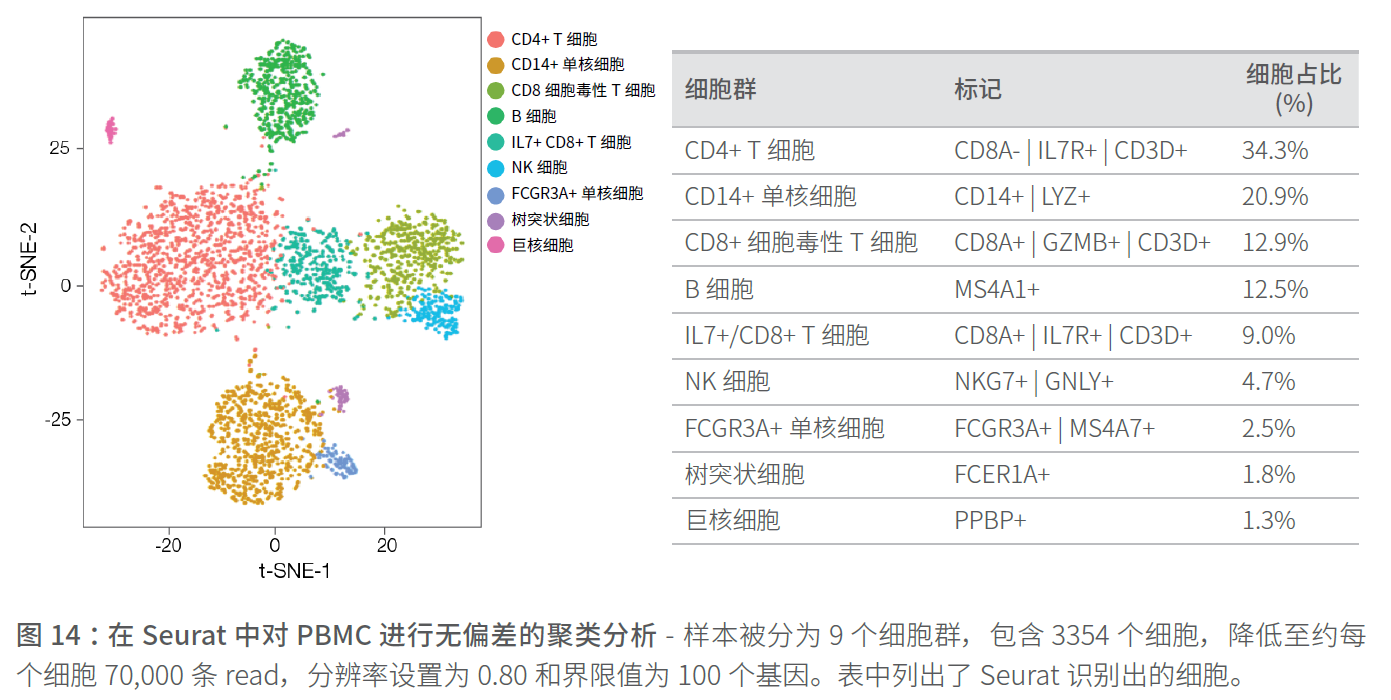
Seurat
Seurat 是一个基于R 语言的单细胞RNA-Seq 分析软件,可使用归一化、降维方法、绘图、热图和数据集成等工具来评估细胞异质性。Seurat 使用了降维方法将多维数据(例如数千个细胞,而且每个细胞都有数千个表达基因)转换为可以理解的形式,通过数学方法将维数减少到二维或三维来表示。由此得到的细胞聚类对应于具有不同特征的特定细胞状态或类型。
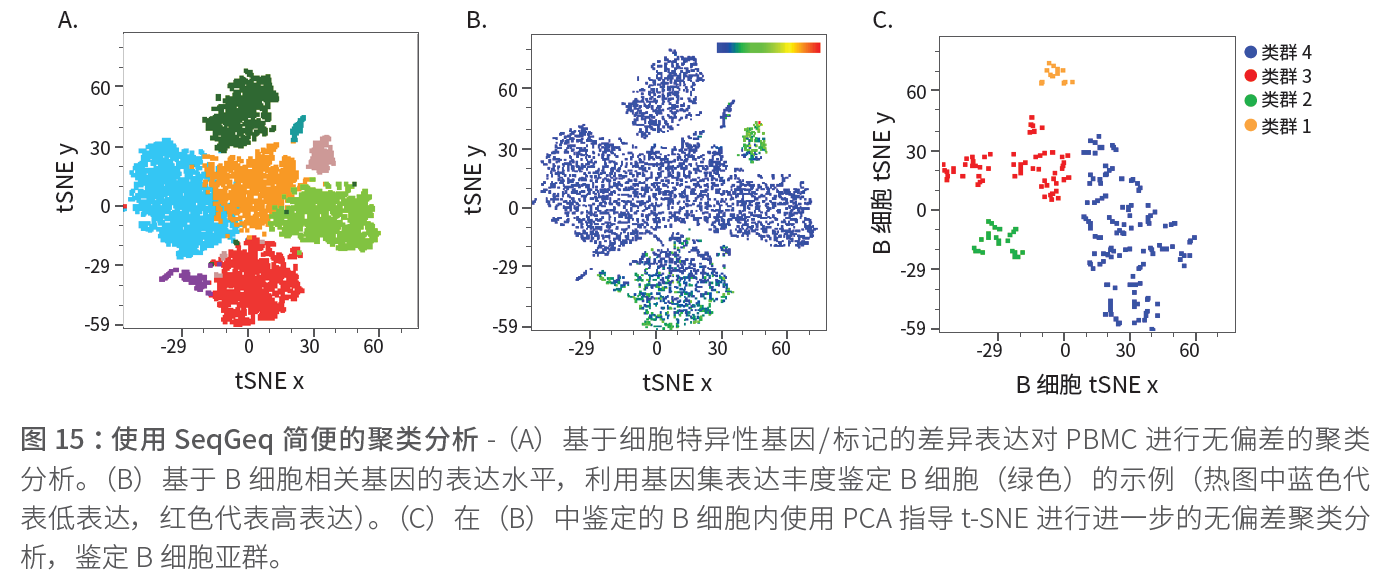
4.6 使用SeqGeq软件进行高级数据可视化
SeqGeq 软件是单细胞基因表达数据的高级数据分析、探索和可视化桌面应用程序,由FlowJo, LLC.(现属于BD Biosciences)开发。SeqGeq 软件具有强大的数据降维和类群识别功能。该软件与BaseSpaceSequence Hub 直接整合,能生成单细胞统计颜色映射图和汇总热图,具有拖拽报告编辑器,能可视化和分析基因表达数据(下图)。