来源:单细胞天地
一、前言
scRNA-seq技术到目前为止也有一百多个了,但主流的可以大致分为以下几种:
-
Droplet-based: 10X Genomics, inDrop, Drop-seq
-
Plate-based with unique molecular identifiers (UMIs): CEL-seq, MARS-seq
-
Plate-based with reads: Smart-seq2
-
Other: sci-RNA-seq, Seq-Well
实际上我们需要理解的就是10x数据和Smart-seq2技术啦,最常用而且最常见!
二、10X Genomics的原始数据
目前单细胞是10x的天下,而10x的测序数据,御用软件cellranger其实就是star的包装,关于10X仪器的单细胞转录组数据走cellranger流程,教程如下:
输入数据是测序序列的fastq文件,输出的是表达矩阵。值得注意的是,每个10x样本都有3个fastq文件作为输入,然后输出的表达矩阵,也是3个文件。
在教程:使用seurat3的merge功能整合8个10X单细胞转录组样本和seurat3的merge功能和cellranger的aggr整合多个10X单细胞转录组对比,也给出了后续R代码读取10x单细胞转录组数据的3个文件的表达矩阵。
三、10X Genomics的单细胞公共数据
比如 GSE128033 和 GSE135893,就是10x数据集,随便下载其中一个,就能看到每个样本都是走流程拿到10x单细胞转录组数据的3个文件的表达矩阵。
2.2M Mar 8 2019 GSM3660655_SC94IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660655_SC94IPFUP_genes.tsv.gz
26M Mar 8 2019 GSM3660655_SC94IPFUP_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660656_SC95IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660656_SC95IPFLOW_genes.tsv.gz
31M Mar 8 2019 GSM3660656_SC95IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660657_SC153IPFLOW_barcodes.tsv.gz
259K Mar 8 2019 GSM3660657_SC153IPFLOW_genes.tsv.gz
33M Mar 8 2019 GSM3660657_SC153IPFLOW_matrix.mtx.gz
2.2M Mar 8 2019 GSM3660658_SC154IPFUP_barcodes.tsv.gz
259K Mar 8 2019 GSM3660658_SC154IPFUP_genes.tsv.gz
31M Mar 8 2019 GSM3660658_SC154IPFUP_matrix.mtx.gz
下游处理的时候,一定要保证这3个文件同时存在,而且在同一个文件夹下面。
示例代码是:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
sce1 <- CreateSeuratObject(Read10X('../10x-results/WT/'),
"wt")
重点就是 Read10X 函数读取 文件夹路径,比如:../10x-results/WT/ ,保证文件夹下面有3个文件。
四、Smart-seq2数据
这个其实就是普通的转录组数据处理流程哦,比如我们看2017-scRNA-seq-primary breast cancer,韩国研究团队是这样描述的:

其实Smart-seq2单细胞最出名的应该是北京大学张泽民教授团队的多种癌症免疫浸润T细胞测序,包括肝癌,结直肠癌,肺癌的,比如 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98638

这样的表达矩阵,每个样本只有一个文件即可,需要注意的是,提供多种多样数据格式,需要自行阅读量过两万的综述翻译及其细节知识点的补充:
上游分析对大家来说比较困难,除非你有Linux基础,代码如下:
hisat2=/home/jianmingzeng/biosoft/HISAT/hisat2-2.0.4/hisat2
# # 如果使用conda安装的 hisat2,那么 hisat2 命令应该是在环境变量的。
## 索引文件需要自己下载
# https://ccb.jhu.edu/software/hisat2/manual.shtml
# wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/mm10.tar.gz
index=/home/jianmingzeng/reference/index/hisat/mm10/genome
ls raw_fq/*gz | while read id; do
$hisat2 -p 10 -x $index -U $id -S ${id%%.*}.hisat.sam
done
ls *.sam|while read id ;do (samtools sort -O bam -@ 5 -o $(basename ${id} ".sam").bam ${id});done
rm *.sam
ls *.bam |xargs -i samtools index {}
## gtf文件推荐去gencode数据库下载
gtf=/home/jianmingzeng/reference/gtf/gencode/gencode.vM12.annotation.gtf
featureCounts=/home/jianmingzeng/biosoft/featureCounts/subread-1.5.3-Linux-x86_64/bin/featureCounts
# # # 如果使用conda安装的 subread,那么featureCounts 命令应该是在环境变量的。
$featureCounts -T 5 -p -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
# 得到的就是 count矩阵
大家可能会觉得奇怪,为什么我给到的代码里面的软件,都不是截图文献里面使用的呢?其实转录组数据处理流派太多了,并没有绝对的权威。
同样的,很大概率你不需要自己处理上游数据,而是公共数据库,比如我通常是下载上面的count矩阵,然后走代码:
rm(list=ls())
options(stringsAsFactors = F)
# install.packages('R.utils')
# install.packages('data.table')
library(data.table)
# 这个表达矩阵其实是10x的,不过可以演示
a=fread('GSE117988_raw.expMatrix_PBMC.csv.gz',header = TRUE)
length(a$V1)
length(unique(a$V1))
hg=a$V1
dat=a[,2:ncol(a)]
rownames(dat)=hg
hg[grepl('^MT-',hg)]
colnames(dat)
rownames(dat)
meta=as.data.frame(colnames(dat))
colnames(meta)=c('cell name')
rownames(meta)=colnames(dat)
head(meta)
## 前面大量的代码,都是数据预处理
library(Seurat)
dat[1:4,1:4]
class(dat)
# 重点是构建 Seurat对象
pbmc <- CreateSeuratObject(counts = dat,
meta.data = meta,
min.cells = 3, min.features = 200,project = '10x_PBMC')
pbmc
head(colnames(dat))
head(rownames(dat))
rownames(GetAssayData(pbmc,'counts'))
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
两种单细胞转录组数据,都是走Seurat流程,只不过是构建 Seurat对象的代码不一样,注意仔细分辨!
五、直接用Seurat读取GEO中的单细胞测序表达矩阵
5.1 常见的单细胞count matrix
- (1)Cell Ranger生成的raw count
Cell Ranger (v3.0)中生成的文件除了bam文件外主要就是如下的三个表格文件(Seurat 的示例文件,2700个pbmc细胞单细胞测序):
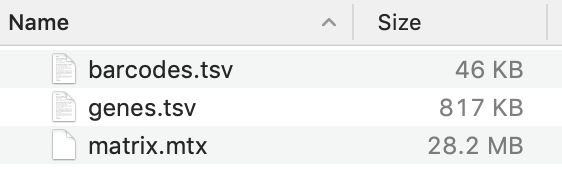
我们可以利用head命令检查数据三个表格的内容。
Barcodes通俗来讲就是每个细胞的代码,组成就是ATCG四个碱基排列组合成的不同的14个碱基组合;
Gene.tsv或者features.tsv一般是基因的ensembl ID 和symbol
matrix.mtx说白了就是每个细胞不同基因的表达矩阵,我们利用分别检查文件的开头和结尾:
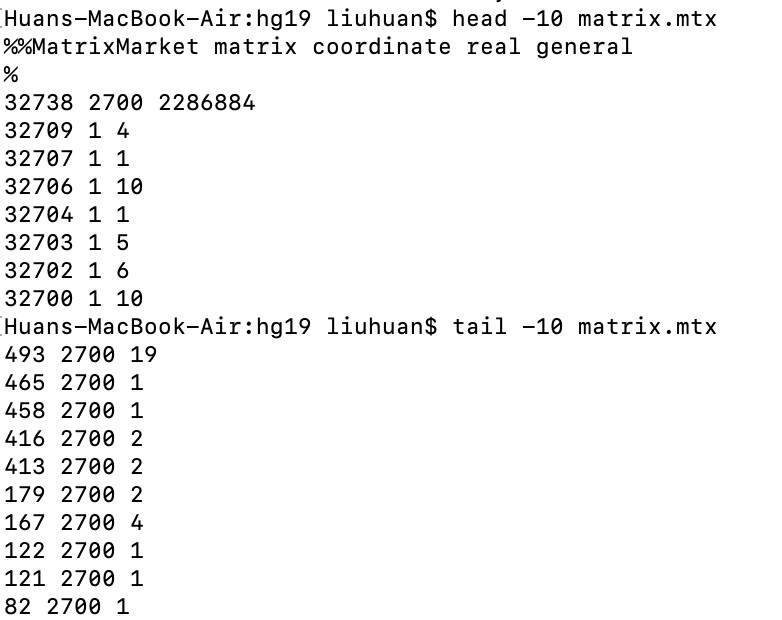
这里我们可以发现其实就是2700个细胞不同基因的表达(第一列是基因的ID,用于与genes.tsv对应转换;第二列则是细胞的编号,匹配barcodes.tsv;第三列则是基因的表达量TPM)(没有表达的基因不做记录)这三组表格组合成。理解这三个表格组成后我们也不难发现,缺一不可的是matrx.mtx文件,而genes.tsv则一般是用于注释的基因组通用文件;而如果缺失barcodes.tsv的话,则可以根据matrix判断细胞数量自己“人为构建出”相应数量不同的barcode表格或者利用samtools从bam文件获取。当我们把这三个文件后存在一个独立文件夹后可以直接利用Seurat (v3.0)的Read10X()命令读取并构建成行名称为基因名,列名称为barcode序列(基因名x细胞)的表达矩阵(也就是SeuratObject)进行后续分析。如果我们只想从这三个表格直接整合成一个(基因名x细胞)的表达矩阵,可以利用以下代码完成:
library(Matrix)
matrix_dir = "~/filtered_feature_bc_matrix/hg19/" ##根据实际文件夹进行修改
barcode.path <- paste0(matrix_dir, "barcodes.tsv")
features.path <- paste0(matrix_dir, "genes.tsv")
matrix.path <- paste0(matrix_dir, "matrix.mtx")
mat <- readMM(file = matrix.path)
feature.names = read.delim(features.path,
header = FALSE,
stringsAsFactors = FALSE)
barcode.names = read.delim(barcode.path,
header = FALSE,
stringsAsFactors = FALSE)
colnames(mat) = barcode.names$V1
rownames(mat) = feature.names$V1
- (2)从公共数据库中获取的count matrix
拿我们常见的GEO数据库为例,如果是上传到GEO数据的数据必须要上传处理后的数据(https://www.ncbi.nlm.nih.gov/geo/info/seq.html),这一方面方便其他研究人员直接更快速的获取或者验证最初的高通量测序,减少了下载SRA粗数据并进行重新比对的时间。
一般来讲这些数据往往是整合好的一个count matrix,比如最新上传的一组造血干细胞单细胞测序数据(A 3D Atlas of Hematopoietic Stem and Progenitor Cell Expansion by Multi-dimensional RNA-Seq Analysis)(GSE120503),我们看到的处理后数据是单个文件,如下图所示:
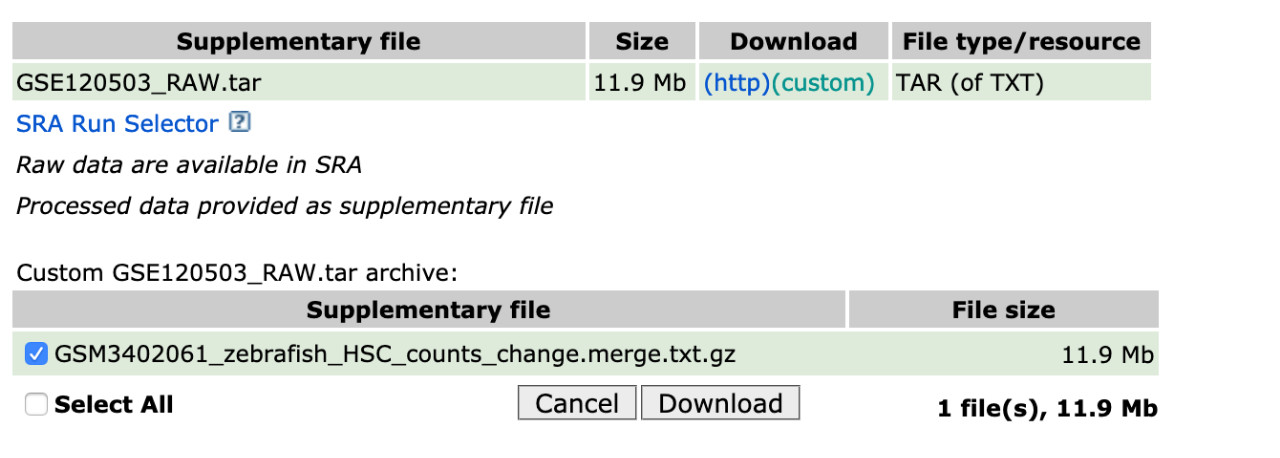
解压后我们得到只有一个叫做“GSM3402061_zebrafish_HSC_counts_change.merge.txt”的文件,而不是Cell Ranger输出的三个文件。
我们检查一下文件的内容:

其实这就是我们在上一步整合出的(基因 x 细胞)的表达矩阵,那么如果我们想直接利用Seurat导入这个表达矩阵进行后续分析该如何做呢?
5.2 Count matrix导入Seurat
对于上述的表达矩阵,我们不能直接使用Seurat的Read10X()函数进行读取,但是要进行后续分析我们可以直接把这个表达矩阵变成SeuratObject。这是一个R读取表格的基本操作:
setwd("/test/") ##注意工作目录
library(Seurat) ##version 3.0
library(dplyr)
new_counts <- read.table(file="/test/GSM3402061_zebrafish_HSC_counts_change.merge.txt")
head(new_counts)
mydata <- CreateSeuratObject(counts = new_counts, min.cells = 3, project = "mydata_scRNAseq")
通过以上两种操作我们就可以完成Cell Ranger产出数据与SeuratObject之间的互相转换。而利用这种简单的几行命令,我们可以较快的从他人上传好的数据中获取我们所需的信息(当然这需要我们充分相信合作者或者数据上传人对于数据处理的数据质量),节省了大量下载和处理数据的时间。
六、Q&A
6.1 很大概率上你并不会需要自己走上游流程
主要是因为对计算资源的消耗,实验室搭建上游流程成本太高,还不如一次性付费让公司做出来表达矩阵给到你后下游慢慢探索。但是懂上游分析流程有助于你更好的认识你的单细胞数据!
6.2 为什么10x单细胞转录组表达矩阵有3个文件
因为10x单细胞转录组表达矩阵里面的0值非常多,所以换成3个文件存储更节省空间。本质上仍然是一个表达矩阵而已,如果你都有了表达矩阵,就没必要去想那3个文件了。
6.3 最少要测多少的样本量和数据量
单个样本的单细胞转录组很少见了,现在以2个样本项目居多,一个对照一个处理,如果是常规转录组,两个分组的话每个组通常是3个样本,但是我们说了嘛,单细胞还是很贵,单个10x样本还是在3万左右的费用,大多数课题组就是想尝个鲜,而且学术界也承认单细胞的确很贵大家hold不住,所以2个样本项目也可以发出去的啦。

如果样本量比较多,复杂的实验设计,不同生物学假设的分组,可以用示意图发方式,如下:
6.4 什么是测序数据量
测序数据量,其实就是文库大小,每个细胞的reads的总数。
上面截图的10x的单细胞转录组的两个样本的课题,就写清楚了,每个样本是3500左右细胞,每个细胞是8万的reads数量。实际上10x单细胞转录组每个样本可以是3000到10000的细胞数量都可以,取决于实验设计。每个细胞平均可以是1到10万的reads,都没有问题。如果是Smart-seq2技术呢,每个样本细胞数量通常是96的倍数,500个左右就很厉害了,然后每个细胞的reads就可以很多,百万级别都没有问题。
6.4 测序数据量和捕获细胞数量对结果的影响
10X官方有PBMC单细胞测试数据,4000K细胞,每个细胞平均是50K的reads。然后取,500, 1k, 2,5k, 5k, 7.5k, 10k, 15k, 25k的随机抽样子集,同样的,取100,200,400,600,500,1,2,3,4K的随机抽样子集。
发现平均每个细胞的0.5K和86K的reads测序量,检测到细胞数量都有4000,而且极低深度测序,仍然是可以比较清晰可见的区分细胞亚型,哪怕在每个细胞的0.5K这样的reads数量情况下每个细胞仅仅是能检测到160个左右的基因。
当然了,这样的探索,仅仅是计算层面的指导,其实你肯定是在公司测序,那么公司的人一定会推荐你每个样本是3~8K细胞,平均每个细胞15-50K的reads,这样的测序策略。