来源:单细胞天地
一、过滤不合格细胞和基因
拿到表达矩阵后有一个很重要的质控过程,就是根据一些阈值来过滤不合格细胞和基因。细胞的不合格很容易理解, 可能是那个细胞检测到的基因数量太少,或者太多,也许是那个细胞的文库大小异常,都是需要谨慎考虑是否管理它。
基因的不合格,就需要大家对人类参考基因组的注释信息有所背景了。走RNA-seq定量流程,拿到的表达矩阵通常是取决于gtf文件的注释程度,人类的gtf里面五万多个基因,不可能都在你的单细胞转录组项目数据里面出现。
1.1 首先,细胞的取舍主要是看文库大小和检测到的基因数量
单细胞转录组通常使用10x数据,所以细胞数量惊人,每个样本可以是3000到10000的细胞数量都可以,取决于实验设计。每个细胞平均可以是1到10万的reads文库,都没有问题。每个细胞可以检测到多少基因数量,就取决于每个细胞分到的reads总数,1到10万的reads文库对应着200到1000的基因数量。
但是Smart-seq2技术的单细胞数据就不一样了,每个样本细胞数量通常是96的倍数,500个左右就很厉害了,然后每个细胞的reads就可以很多,百万级别都没有问题。所以检测到基因数量也很多,如下:

所以在你设置阈值来过滤不合格细胞的时候,一定要先理解好你的单细胞转录组数据,给全部的细胞检测到的基因数量绘制一个boxplot,看看哪些细胞所检测到的基因数量偏多或者偏少,一般来说,这样的的离群细胞就是你需要去除的。代码如下:
box <- lapply(colnames(sample_ann[,1:19]),function(i) {
dat <- sample_ann[,i,drop=F]
dat$sample=rownames(dat)
## 画boxplot
ggplot(dat, aes('all cells', get(i))) +
geom_boxplot() +
xlab(NULL)+ylab(i)
})
plot_grid(plotlist=box, ncol=5 )
这样的话细胞的上游指标可以批量检验,如下:
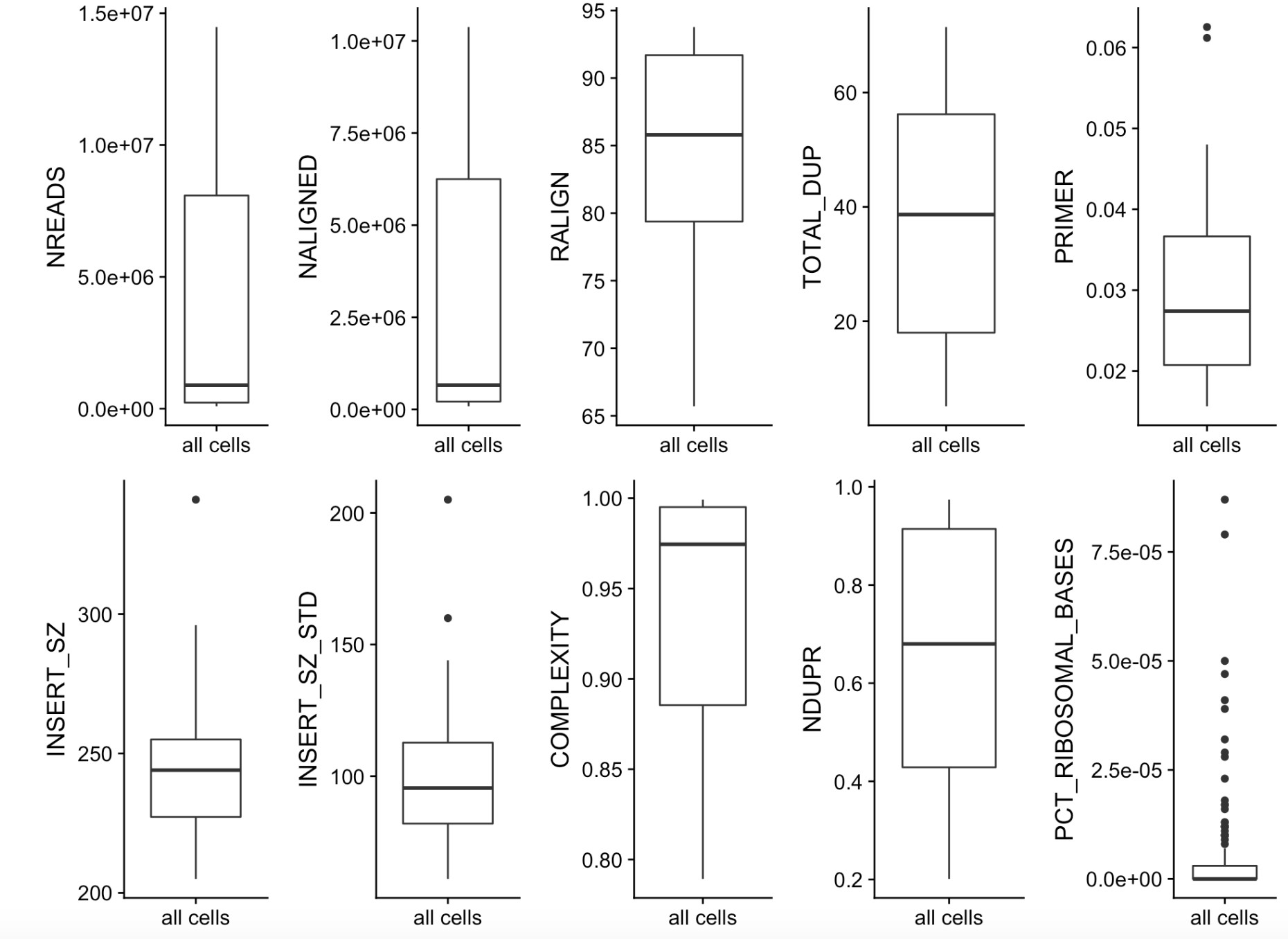
可以看到,每个细胞的测序文库大小和检测到基因数量虽然很重要,上游分析的其它质量指标,比对率,质量分数,duplicate情况,插入片段,内含子误差其实也会影响这个细胞的取舍。
1.2 然后,删除一些基因
前面我们提到过,gtf文件的注释程度不一样,拿到的表达矩阵通常是全部的基因,比如人类的gtf里面五万多个基因,你的表达矩阵就是5万多行。
实际上,大量的基因在你所有的细胞里面都是不表达的,这就是表达量全部为0的,肯定是需要去除啦。还有一些基因仅仅是在5%左右的细胞表达,这个时候就很主观了,可能确实这个基因是那5%的稀有细胞的marker基因,也有可能就是单细胞转录组技术导致这个基因大量的drop-out了。
这个时候,你就需要卡一个阈值,到底一个基因表达多少算是表达呢,到底一个基因在多少个细胞里面有表达,你才保留下去,做下游分析。当然了,过滤细胞或者基因仅仅是单细胞转录组项目质量控制的开始,后面还有线粒体基因含量,核糖体基因含量,高表达量基因比例,细胞周期,批次效应。
二、过滤线粒体核糖体基因
我们埋下了一个伏笔,就是拿到了差不多干净的表达矩阵后还有一个预处理步骤,但是这个步骤是选修,就是说你不会过滤线粒体核糖体基因也可以不做,没有人会责怪你,因为单细胞转录组数据分析本来就有难度。分析要点无穷无尽,即使你真的准备过滤线粒体核糖体基因你也会发现标准不好把握,需要看很多文献,还得结合你自己项目数据的实际情况啦。而且过滤线粒体核糖体基因并不是质控的终点,你还有细胞周期检查,单细胞活性检查,是否有两个细胞混在一起也是需要检查,非常的累。不仅仅是标准不好把握,而且因项目而异,具体情况具体判断。
2.1 处理线粒体核糖体基因的几个策略
2.1.1 粗暴的去除全部线粒体核糖体基因
直接在表达矩阵里面,去除掉属于的那一行表达量即可,有点类似于甲基化芯片数据分析,直接去除性染色体上面的全部探针,如下所示:

2.1.2 选择一个阈值来去除高表达量细胞
如下所示:

这个阈值很大程度上取决于你对自己项目的了解程度,不同器官组织提取的单细胞,本来就线粒体基因平均水平不一样, 不能一刀切!
这个只能是靠你看文献来获取认知了,多看多学习!
我的经验是,多次反复查看线粒体核糖体基因的影响,分群前后看,不同batch看,多次质控图表里面显示它,判断它是否是一个主要因素。
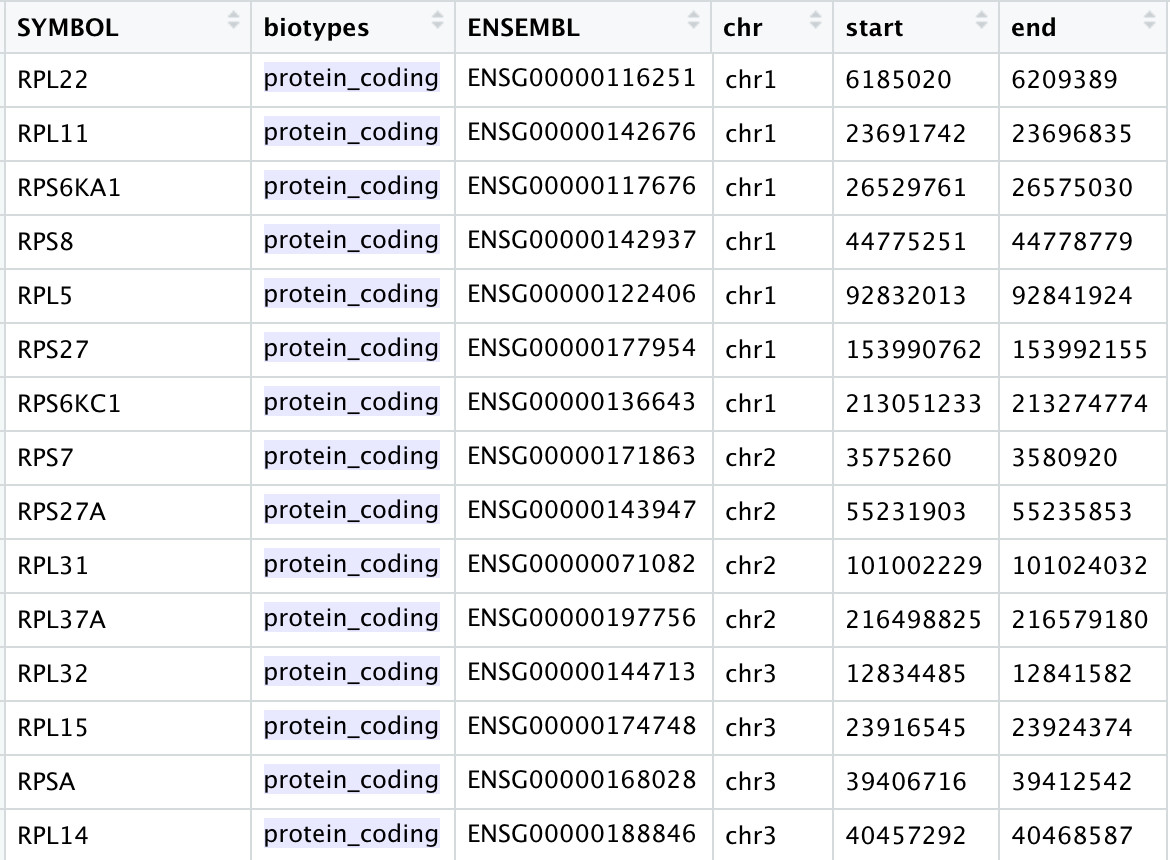
2.2 人类和小鼠的线粒体核糖体基因有哪些
以gencode数据库的gtf文件为标准,在人类的gencode.v32.annotation.gtf 文件里面,可以查找到37个:
gene_id "ENSG00000210049.1"; gene_name "MT-TF"; hgnc_id "HGNC:7481"
gene_id "ENSG00000211459.2"; gene_name "MT-RNR1"; hgnc_id "HGNC:7470"
gene_id "ENSG00000210077.1"; gene_name "MT-TV"; hgnc_id "HGNC:7500"
gene_id "ENSG00000210082.2"; gene_name "MT-RNR2"; hgnc_id "HGNC:7471"
gene_id "ENSG00000209082.1"; gene_name "MT-TL1"; hgnc_id "HGNC:7490"
gene_id "ENSG00000198888.2"; gene_name "MT-ND1"; hgnc_id "HGNC:7455"
gene_id "ENSG00000210100.1"; gene_name "MT-TI"; hgnc_id "HGNC:7488"
gene_id "ENSG00000210107.1"; gene_name "MT-TQ"; hgnc_id "HGNC:7495"
gene_id "ENSG00000210112.1"; gene_name "MT-TM"; hgnc_id "HGNC:7492"
gene_id "ENSG00000198763.3"; gene_name "MT-ND2"; hgnc_id "HGNC:7456"
gene_id "ENSG00000210117.1"; gene_name "MT-TW"; hgnc_id "HGNC:7501"
gene_id "ENSG00000210127.1"; gene_name "MT-TA"; hgnc_id "HGNC:7475"
gene_id "ENSG00000210135.1"; gene_name "MT-TN"; hgnc_id "HGNC:7493"
gene_id "ENSG00000210140.1"; gene_name "MT-TC"; hgnc_id "HGNC:7477"
gene_id "ENSG00000210144.1"; gene_name "MT-TY"; hgnc_id "HGNC:7502"
gene_id "ENSG00000198804.2"; gene_name "MT-CO1"; hgnc_id "HGNC:7419"
gene_id "ENSG00000210151.2"; gene_name "MT-TS1"; hgnc_id "HGNC:7497"
gene_id "ENSG00000210154.1"; gene_name "MT-TD"; hgnc_id "HGNC:7478"
gene_id "ENSG00000198712.1"; gene_name "MT-CO2"; hgnc_id "HGNC:7421"
gene_id "ENSG00000210156.1"; gene_name "MT-TK"; hgnc_id "HGNC:7489"
gene_id "ENSG00000228253.1"; gene_name "MT-ATP8"; hgnc_id "HGNC:7415"
gene_id "ENSG00000198899.2"; gene_name "MT-ATP6"; hgnc_id "HGNC:7414"
gene_id "ENSG00000198938.2"; gene_name "MT-CO3"; hgnc_id "HGNC:7422"
gene_id "ENSG00000210164.1"; gene_name "MT-TG"; hgnc_id "HGNC:7486"
gene_id "ENSG00000198840.2"; gene_name "MT-ND3"; hgnc_id "HGNC:7458"
gene_id "ENSG00000210174.1"; gene_name "MT-TR"; hgnc_id "HGNC:7496"
gene_id "ENSG00000212907.2"; gene_name "MT-ND4L"; hgnc_id "HGNC:7460"
gene_id "ENSG00000198886.2"; gene_name "MT-ND4"; hgnc_id "HGNC:7459"
gene_id "ENSG00000210176.1"; gene_name "MT-TH"; hgnc_id "HGNC:7487"
gene_id "ENSG00000210184.1"; gene_name "MT-TS2"; hgnc_id "HGNC:7498"
gene_id "ENSG00000210191.1"; gene_name "MT-TL2"; hgnc_id "HGNC:7491"
gene_id "ENSG00000198786.2"; gene_name "MT-ND5"; hgnc_id "HGNC:7461"
gene_id "ENSG00000198695.2"; gene_name "MT-ND6"; hgnc_id "HGNC:7462"
gene_id "ENSG00000210194.1"; gene_name "MT-TE"; hgnc_id "HGNC:7479"
gene_id "ENSG00000198727.2"; gene_name "MT-CYB"; hgnc_id "HGNC:7427"
gene_id "ENSG00000210195.2"; gene_name "MT-TT"; hgnc_id "HGNC:7499"
gene_id "ENSG00000210196.2"; gene_name "MT-TP"; hgnc_id "HGNC:7494"
所以拿到自己的表达矩阵后,其实简单的看看基因名字是否以 MT- 开头即可哦。所以会看到seurat包的函数:PercentageFeatureSet 可以用来计算线粒体基因含量。
sce <- CreateSeuratObject(Read10X('../scRNA/filtered_feature_bc_matrix/'), "sce")
head(sce@meta.data)
GetAssayData(sce,'counts')
sce[["percent.mt"]] <- PercentageFeatureSet(sce, pattern = "^MT-")
VlnPlot(sce, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
上面的方法是修改 sce[[“percent.mt”]] ,下面我们演示 AddMetaData 函数,同样是可以增加线粒体基因含量信息到我们的seurat对象。
mt.genes <- rownames(sce)[grep("^MT-",rownames(sce))]
C<-GetAssayData(object = sce, slot = "counts")
percent.mito <- Matrix::colSums(C[mt.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.mito, col.name = "percent.mito")
sce[["percent.mito"]]
也可以是添加核糖体基因含量,同样的你需要知道核糖体基因的名字规则:
rb.genes <- rownames(sce)[grep("^RP[SL]",rownames(sce))]
percent.ribo <- Matrix::colSums(C[rb.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.ribo, col.name = "percent.ribo")
在人类的gencode.v32.annotation.gtf 文件里面,可以看到核糖体基因数量也不少哦。
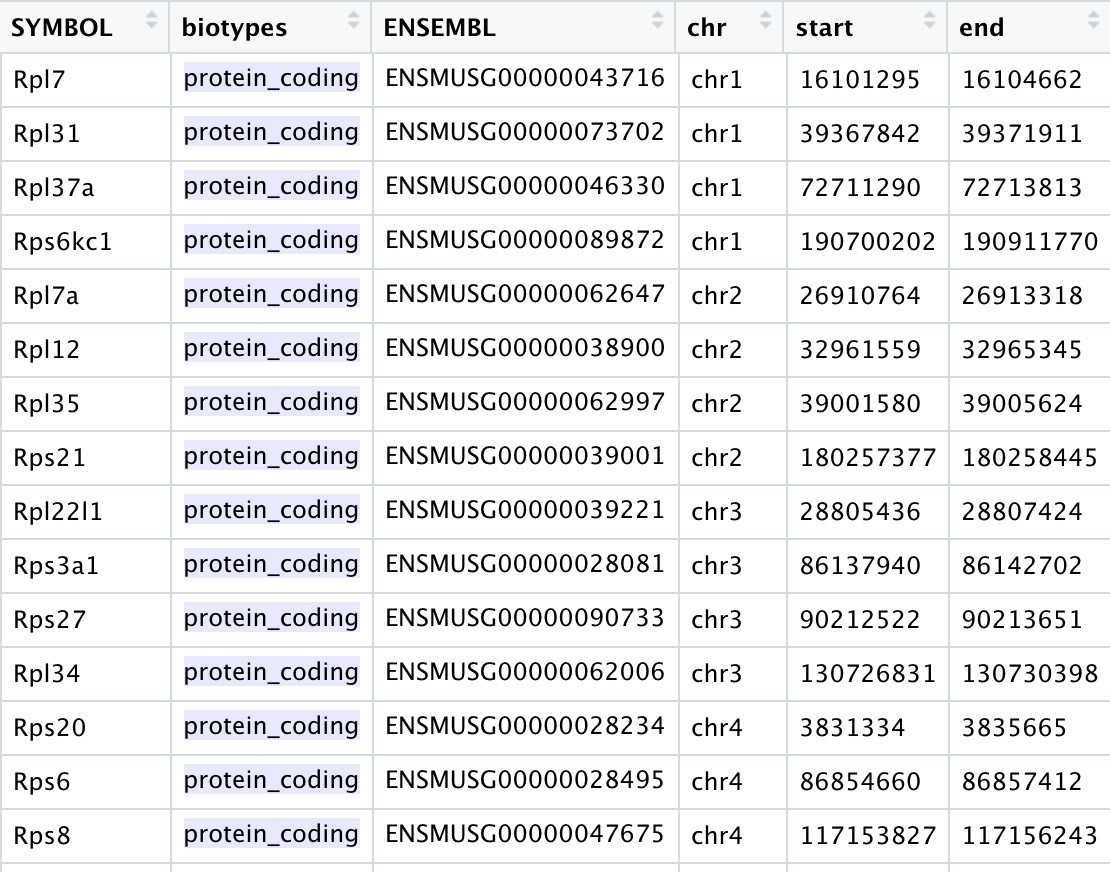
如果是小鼠,通常是基因名字大小写替换一下:
2.3 不仅仅是线粒体核糖体基因
还可以去免疫相关基因,缺氧相关基因,就更加的需要深入到你自己的课题,其实细节是无穷无尽的,但是我们的教学没办法做到如此的个性化,只能是精炼了常规单细胞转录组数据分析主线,就是5大R包, scater,monocle,Seurat,scran,M3Drop,然后10个步骤:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因进行亚群命名
- step10: 继续分类
三、去除细胞效应和基因效应
其实质量控制三部曲,还有一个很关键的点没有讲解,就是多个样本整合,并且区分批次效应和生物学差异。但是这个点很大程度是依赖于经验,就是说,要想搞清楚,需要写很多自定义的代码去一点一滴的探索,而不仅仅是流程。所以我们就只能介绍到这里,假设大家都拿到了干净的表达矩阵,而且可以很肯定的说这个表达矩阵做下游分析是ok的。
那么我们就来看看,有了干净的表达矩阵后下游分析的第一个分析要点就是:归一化和标准化。
3.1 归一化和标准化的区别
实际上口语里面通常是没办法很便捷的区分这两个概念,我查阅了大家的资料,发现基本上都是混淆在z-score转换和0-1转换这两个上面。所以我这里把归一化和标准化替换成为去除样本/细胞效应或者去除基因效应:
- 首先去除样本/细胞效应因为不同样本或者细胞的测序数据量不一样,所以同样的一个基因在不同细胞,哪怕你看到的表达量是一样的,但是背后细胞整体测序数据量的差异其实反而说明了这个基因在不同细胞表达量其实是有差异的。在seurat3里面的代码是:
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)
默认文库大小是1万,默认会取log值。
- 然后去除基因效应:这个主要是在绘制热图的时候会需要使用,因为个别基因表达量超级高,在热图里面一枝独秀,实际上我们并不会关心不同基因的表达量高低,我们仅仅是想看指定基因在不同细胞的高低而已,这样的话,就把该基因的表达量在不同细胞的数值,进行z-score这样的转换。在seurat3里面的代码是:
sce <- ScaleData(sce)
这样处理后的表达矩阵,就可以进行后续的降维聚类分群啦。
3.2 取log让表达量离散程度降低
这个时候眼尖的朋友其实看到了,在使用NormalizeData函数的时候,有一个 normalization.method = “LogNormalize” 参数被设置了,这个是为什么呢?
其实很简单,原始的raw counts矩阵是一个离散型的变量,离散程度很高。有的基因表达丰度比较高,counts数为10000,有些低表达的基因counts可能10,甚至在有些样本中为0。因为是单细胞转录组,drop-out现象很严重,其实大量的基因在很多细胞的表达量都是0,如果是10x数据,甚至会出现一个表达矩阵里面97%的数值都是0 的现象。
如果对表达量取一下log10,发现10000变成了4,10变成了1,这样之前离散程度很大的数据就被集中了。
有时当表达量为0时,取log会出现错误,可以log(counts+1)来取log值。当x=1时,所有的log系列函数值都为0。这样原本表达量为0的值,取log后仍为0。
这也就是UCSC的XENA下载到的表达矩阵的形式。
3.3 使用GetAssayData函数查看不同形式的表达矩阵
在seurat3里面,很方便进行各种形式的归一化或者标准化,同样的,也很容易查看处理前后的表达矩阵,使用GetAssayData函数即可。
This function can be used to pull information from any of the slots in the Assay class. For example, pull one of the data matrices("counts", "data", or "scale.data").
# 最原始数据
GetAssayData(sce,'counts')[1:6,1:6]
# 去除了细胞测序数据量后
1/(colSums(as.data.frame(GetAssayData(sce,'counts')))[2]/10000)
GetAssayData(sce,'data')[1:6,1:6]
# z-score后
GetAssayData(sce,'scale.data')[1:6,1:6]
结果如下:
> # 最原始数据
> GetAssayData(sce,'counts')[1:6,1:6]
6 x 6 sparse Matrix of class "dgCMatrix"
6A-11 6A-13 6A-14 6A-15 6A-16 6A-17
DDX11L1 . . . . . .
WASH7P . 1 . . . .
MIR6859-1 . . . . . .
RP11-34P13.3 . . . . . .
OR4G11P . . . . . .
OR4F5 . . . . . .
> # 去除了细胞测序数据量后
> 1/(colSums(as.data.frame(GetAssayData(sce,'counts')))[2]/10000)
6A-13
0.009725971
> GetAssayData(sce,'data')[1:6,1:6]
6 x 6 sparse Matrix of class "dgCMatrix"
6A-11 6A-13 6A-14 6A-15 6A-16 6A-17
DDX11L1 . . . . . .
WASH7P . 0.009678978 . . . .
MIR6859-1 . . . . . .
RP11-34P13.3 . . . . . .
OR4G11P . . . . . .
OR4F5 . . . . . .
> # z-score后
> GetAssayData(sce,'scale.data')[1:6,1:6]
6A-11 6A-13 6A-14 6A-15 6A-16 6A-17
RP5-857K21.9 -0.8302953 -0.8302953 -0.7327066 -0.7224892 -0.7683106 -0.7692732
RP5-857K21.8 -0.5066862 -0.5688591 -0.5349974 -0.4881690 -0.3650311 -0.4524682
PRKCZ -0.8350477 -0.5996852 -0.8350477 -0.8350477 -0.5988844 -0.8350477
TNFRSF14 -0.2550752 -0.2550752 -0.2550752 -0.2550752 -0.2550752 -0.2550752
TP73 -0.2761133 -0.2761133 -0.2761133 -0.2761133 -0.2761133 -0.2337521
HES2 -0.4569104 -0.4569104 -0.4569104 -0.4569104 -0.4569104
3.4 其实样本/细胞效应不仅仅是文库大小
每个细胞测序数据量的不一致是很容易理解的,但其实细胞之间还有很多其它效应,比如线粒体基因含量,ERCC含量等等,那些处理起来,其实就是深入了解我们讲解seurat里面的NormalizeData和ScaleData函数。