来源:单细胞天地
前面学习到单细胞转录组数据的降维聚类分群,基本上跑的都是标准代码,里面很多细节参数是需要自己慢慢摸索的。保证单细胞转录组表达矩阵质量ok啦,而且需要去除了各种混杂因素。
因为参数需要自己摸索和调整,所以其实拿到细胞亚群数量是因人而异的,取决于你前面降维的程度,分群的算法和参数。不过最重要的是拿到了不同细胞亚群后需要对它进行命名,给出生物学的解释。不同的人分析同一个数据集,有略微不同的结果是可以接受的,保证自己的生物学故事圆满即可。
1.细胞亚群注释依赖标记基因
文章《Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA》, 就是一个使用seurat标准流程对PBMC分群后如下:
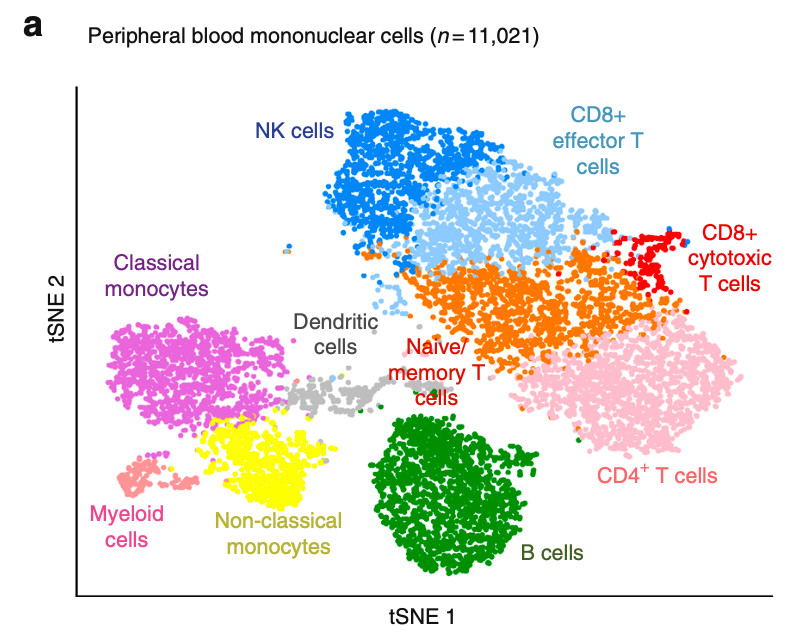
得到了这13个细胞类群,而且也细致的决定它们的亚群名字。实际上PBMC的不同细胞亚群的标记基因是比较明确的,比如在文章:Nucleic Acids Res. 2018 Apr ,dropClust: efficient clustering of ultra-large scRNA-seq data.就写的很清楚:
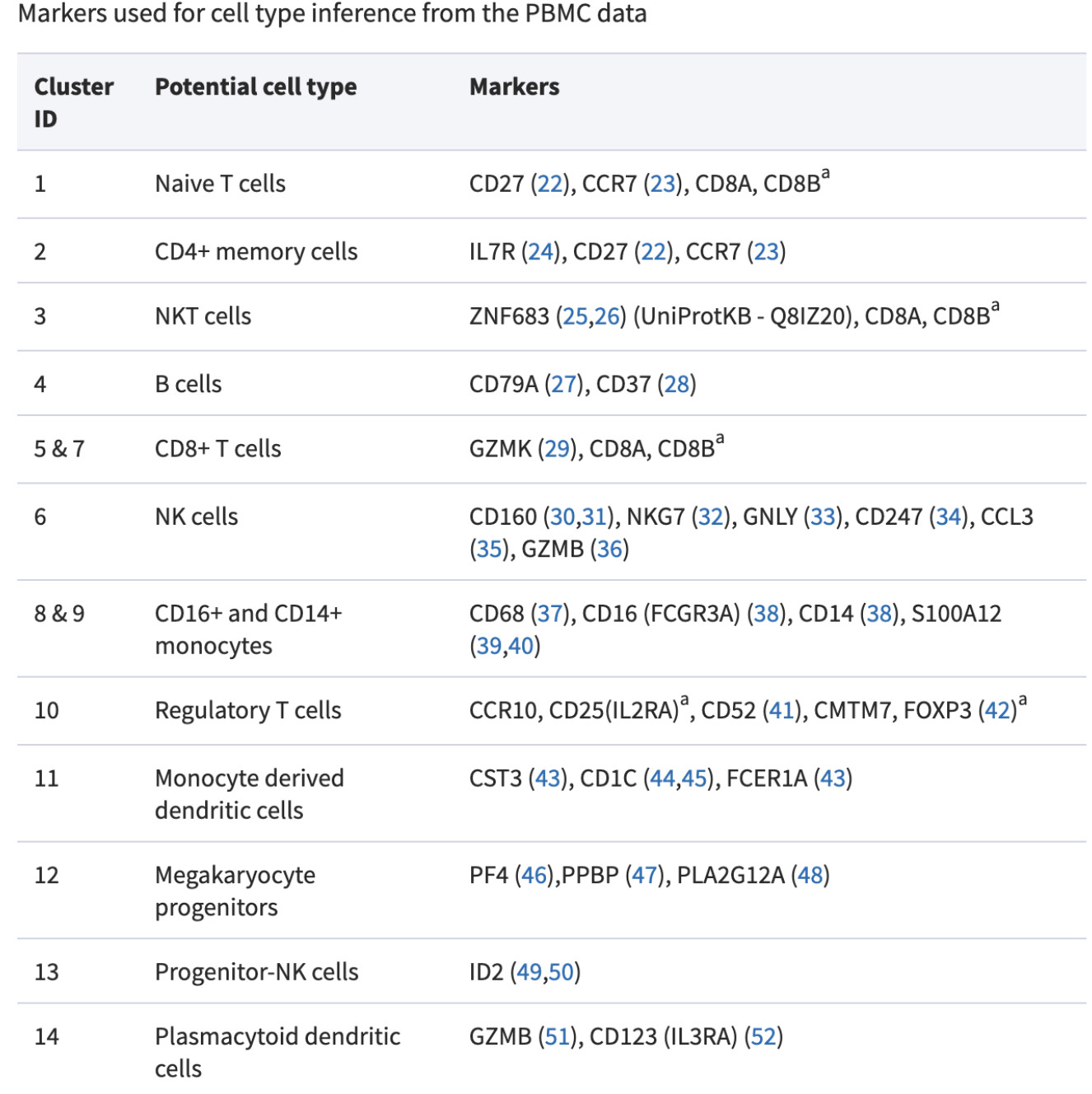
这些标记基因在不同亚群细胞的表达量热图或者小提琴图展示一下,就明白了为什么它们可以作为标记基因,来对细胞亚群进行命名啦。该系列教程如下:
- 10X scRNA免疫治疗学习笔记1-前言
- 10X scRNA免疫治疗学习笔记-2-配置Seurat的R语言环境
- 10X scRNA免疫治疗学习笔记-3-Seurat标准流程
- 10X scRNA免疫治疗学习笔记-4-细胞亚群的生物学命名
- 10X scRNA免疫治疗学习笔记-5-差异分析及可视化
- 10X scRNA免疫治疗学习笔记-6-marker基因的表达量可视化
- 10X scRNA免疫治疗学习笔记-7-条条道路通罗马—单细胞分群分析
如果你看完这些教程,就应该知道,并不是所有你拿到细胞亚群,都是有明确生物学功能的,你的文章所要讲述的生物学故事也并不需要把全部的细胞亚群一一记流水账。
2.标记基因的不同来源
这样的标记基因列表,有一些网页工具会收集,比如cellmarker:
CellMarker: a manually curated resource of cell markers ,作者:X Zhang - 2019 。
这些就需要自行学习了,也有自己查询自己领域内的全部文献, 然后整理出来标记基因列表。这个步骤至少耗时2个月,比如2018年发表于science杂志的文章就是自己根据文献进行整理的:
Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors.
3.也可以通过差异基因来进行注释
比如下面的描述,取top100的差异基因,即便是如此,研究者也仍然是找到了不少已知的标记基因来可视化验证。

4.单个标记基因可视化以小提琴图和热图展现
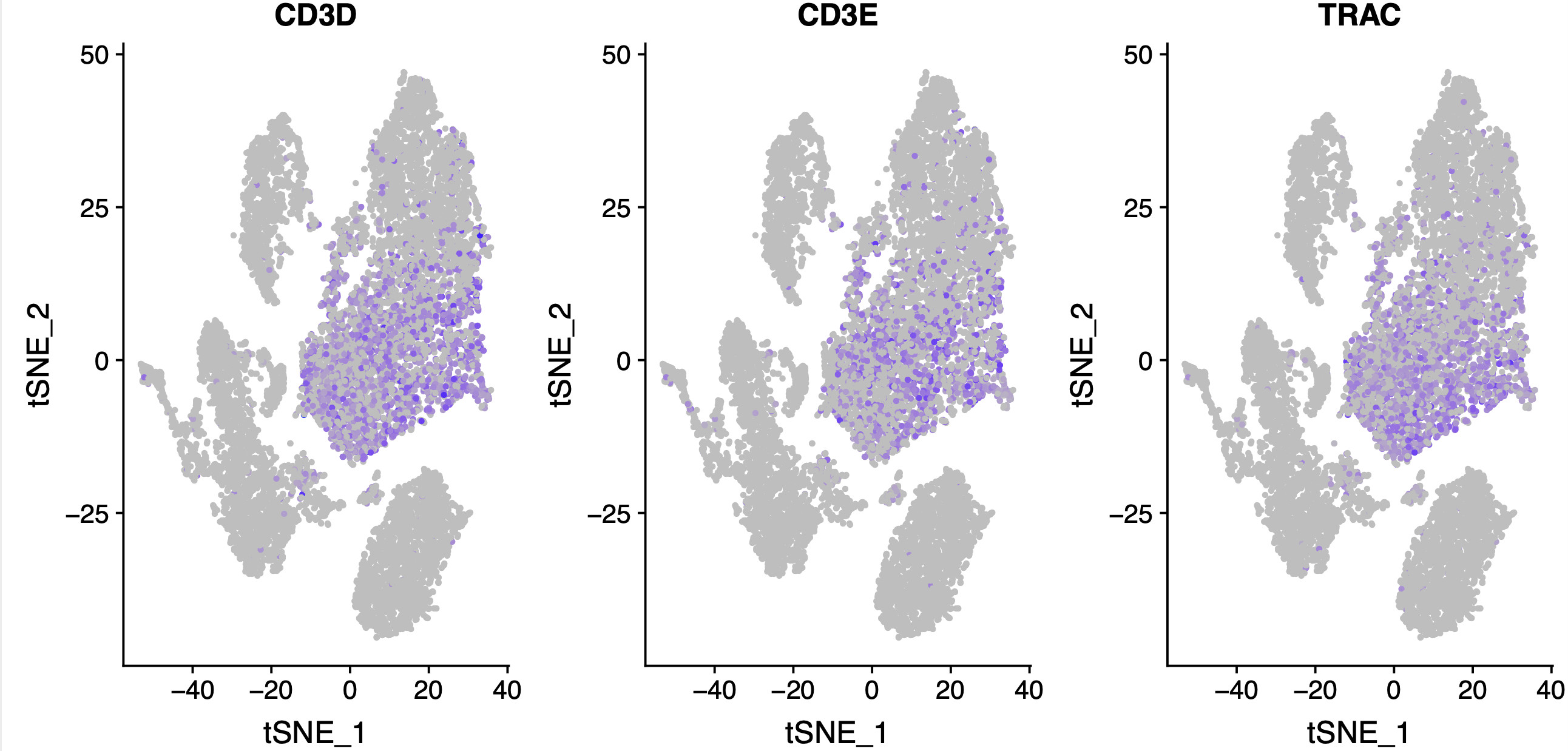
代码如下:
markers_df <- FindMarkers(object = sce, ident.1 = 8, min.pct = 0.25)
print(x = head(markers_df))
markers_genes = rownames(head(x = markers_df, n = 5))
VlnPlot(object = sce, features =markers_genes,log =T )
FeaturePlot(object = sce, features=markers_genes )
就是 VlnPlot 和 FeaturePlot 两个函数,当然,也是可以自己获取细胞的二维平面坐标,以及指定基因的表达值,自定义绘图函数。
5.多个标记基因的可视化也是热图为主
下面的例子是,把细胞亚群的基因表达值取一个统计量(平均值或者中位值)来作为该基因在该细胞亚群的表达量。所以热图展现多个标记基因在全部细胞亚群的表达量热图,也不会拥挤:
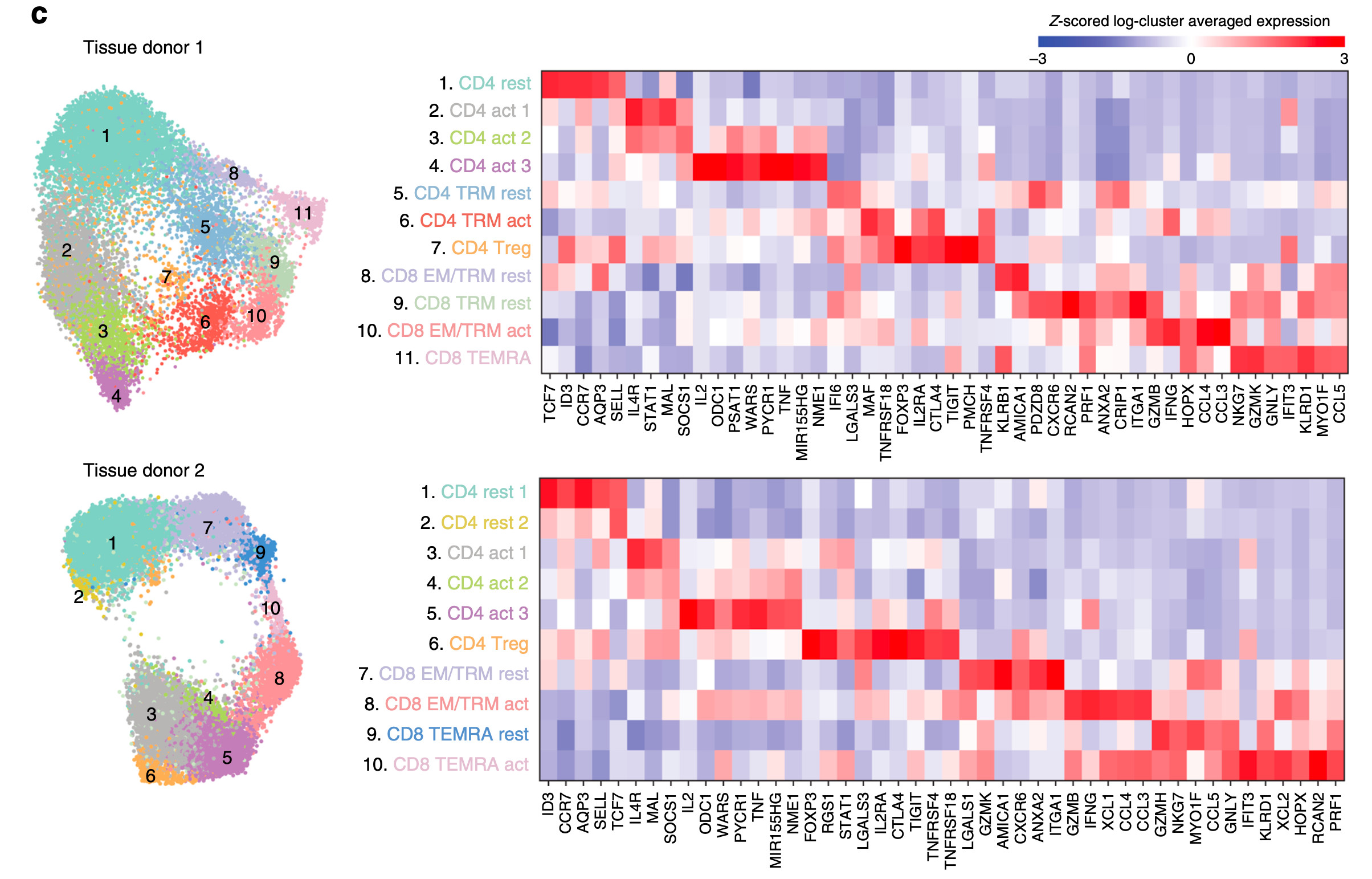
但是,如果要展现多个标记基因在全部细胞的表达量,而不仅仅是细胞亚群,热图就会很拥挤,如下所示:
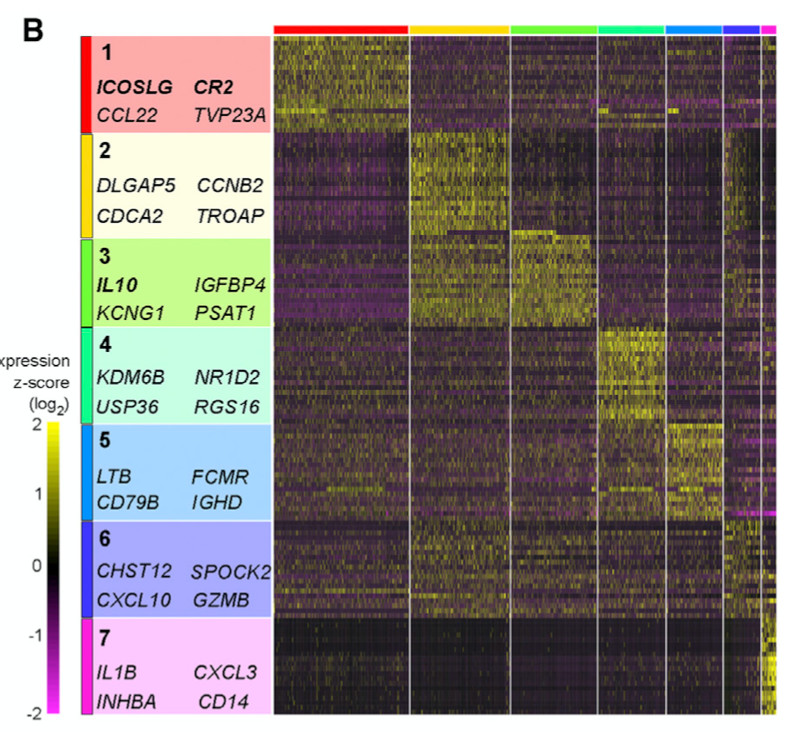
可以看到,每个细胞亚群虽然从注释的角度来说,指定的几个高表达量基因即可定义它,但是它本身特异性高表达的其它基因也会很多。所以作者单独把值得一提的标记基因,在热图坐标高亮出来。