参考资料:Digital-LI
1.SNP的本质属性是什么?什么是allel frequency in population?
SNP是进化过程中随机产生的单点突变,并能稳定地在群体中遗传。其广义上讲是变异:most common type of genetic variation,平级的还有indel、CNV、SV。狭义上讲是标记:biological markers,应为SNP是单碱基的,所以SNP又是一个locus,标记了染色体上的一个位置。大部分人的基因组,99%都是一模一样的,还有些SNP的位点,就是一些可变的位点,在人群中有差异。这些差异/标记可以用于疾病的分析,根据统计学原理,找出与疾病最相关的位点,从而确定某个疾病的risk allele。
allel frequency in population:每一个genome位点都有两个或多个allel,不同allel之间有明显的频率上的差异,简单点理解就是A和a两个性质的频率,但这里是碱基位点,而不是性状基因。
2.GWAS的原理及分析前提
关联分析,就是AS的中文,全称是GWAS。应用基因组中数以百万计的单核苷酸多态;SNP为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。在全基因组范围内选择遗传变异进行基因分析,比较异常和对照组之间每个遗传变异及其频率的差异,统计分析每个变异与目标性状之间的关联性大小,选出最相关的遗传变异进行验证,并根据验证结果最终确认其与目标性状之间的相关性。
核心就是SNP与表型的关联,对于每一个genome位点,如果SNP与phenotype这两个维度协同变化,那我们就可以推测这个SNP极有可能与此phenotype(疾病)相关。简而言之,就是看某个SNP在case和control两个population间是否有allel frequency的显著差异。而现实情况是,我们样本数有限,而且有时候control和case样本不平衡,样本还分男女、人群,而且我们需要对3亿个碱基位点都做统计检验。GWAS的分析工具为PLINK。
GWAS分析的前提:sample size足够,学过统计的都知道sample size会影响power,没有足够的power是得不出正确结论的。GWAS通常需要大量的样本,几千是标配,几百就太少,现在有的都达到了几万几十万级别。一个大误区就是GWAS会测全基因组WGS,其实不是的,那太贵了,大部分是做DNA chip(专业点叫SNP array),只包含了常见的10^6个SNP。稍微有钱的就会上WES,就会得到所有编码区的SNP;最有钱的就是WGS了,全部检测,编码非非编码,比如1000genome计划。
3.曼哈顿图(Manhattan plot)是什么?
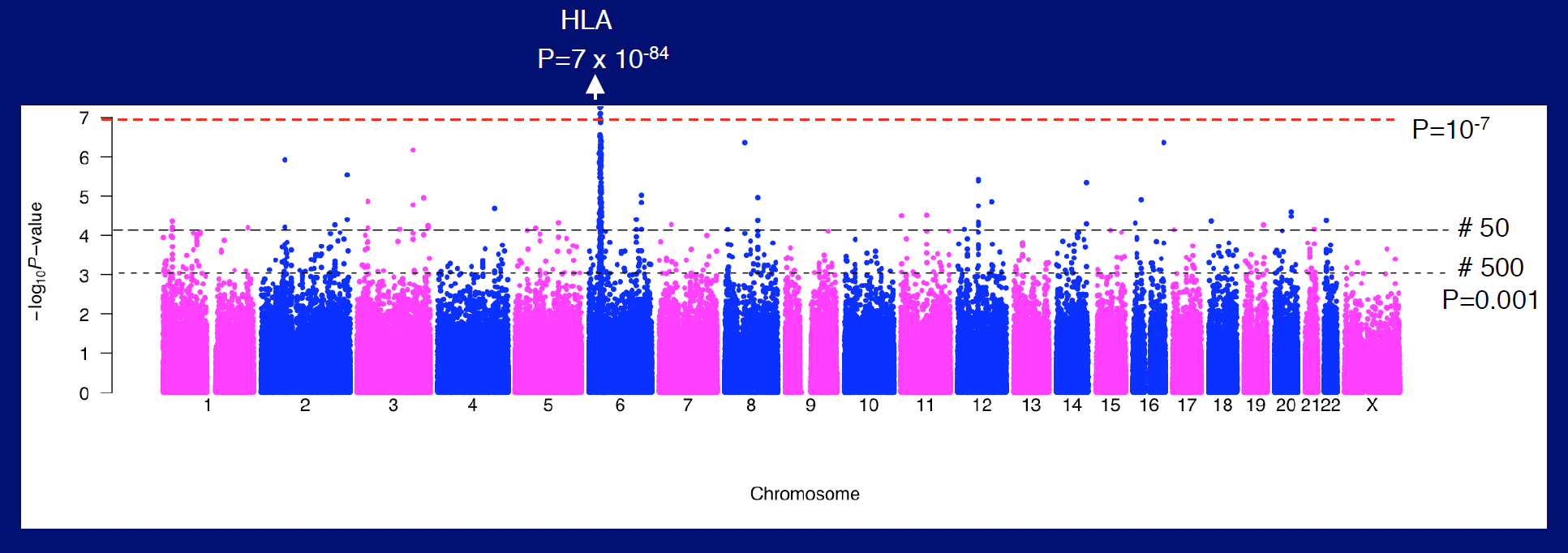
曼哈顿图中的点代表的不是样品,而是SNP。
在生物和统计学上,做频率统计、突变分布、GWAS关联分析的时候,我们经常会看到一些非常漂亮的manhattan plot,能够对候选位点的分布和数值一目了然。位点坐标和pvalue。map文件至少包含三列——染色体号,SNP名字,SNP物理位置。assoc文件包含SNP名字和pvalue。haploview即可画出。
曼哈顿图中,显著的SNP并不是鹤立鸡群的冒出来,而是似乎被捧出来的,就像高楼大厦一样,从底下逐步冒出来的。这一座大厦其实就是连锁在一起的SNP,具有很高的LD score。虽然曼哈顿图里每个点是SNP,但是通常都会把最显著的SNP指向某个基因,因为大家最关注的还是SNP的致病根源,但这样找出来的只有编码区的SNP。
4.什么是fine-mapping?
最突出的SNP极有可能不是causal SNP,它只是near the causal SNP。那么如何找causal SNP呢?fine-mapping
fine mapping:GWAS找到的大多不是causal variants,fine mapping就是为了fill这个gap。GWAS得到大体的SNP后,必须做两方面的深入分析:
- 第一步:对SNP给一个概率上的causality,这就是fine-mapping;
- 第二步:根据功能注释来确定该SNP与可能导致疾病风险改变的基因联系起来。
5.什么是连锁不平衡?
注:GWAS的核心就是连锁不平衡(Linkage disequilibrium,LD),目前大部分的GWAS都是测的array,因为便宜。GWAS会漏掉很多点,所以才会有fine-mapping,根据haplotype来做一些imputation。
连锁不平衡(Linkage disequilibrium,LD):不同基因座的各等位基因在人群中以一定的频率出现。在某一群体中,不同座位某两个等位基因出现在同一条染色体上的频率高于预期的随机频率的现象。(就是孟德尔的分离不是随机的,在染色体上越靠近的allel越倾向于绑在一起,属于物质性的限制。)
例如,两个相邻的基因A和B,它们各自的等位基因为a和b。假设A和B相互独立遗传,则后代群体中观察得到的单倍体基因型AB中出现的P(AB)的概率为P(A)*P(B)。实际观察得到群体中单倍体基因型AB同时出现的概率为P(AB)。计算这种不平衡的方法为:D=P(AB)-P(A)XP(B)。
不连锁就独立,如果不存在连锁不平衡——相互独立,随机组合,实际观察到的群体中单倍体基因型 A和B 同时出现的概率。P (AB) = D + P (A) x P (B) 。D是表示两位点间LD程度值。
事实上,可以检测遍布基因组中的大量遗传标记位点SNP,或者候选基因附近的遗传标记来寻找到因为与致病位点距离足够近而表现出与疾病相关的位点,这就是等位基因关联分析或连锁不平衡定位基因的基本思想。
6.GWAS的分析工具–PLINK
PLINK的主要功能:数据处理、质量控制的基本统计、群体分层分析、单位点的基本关联分析、家系数据的传递不平衡检验、多点连锁分析、单倍体关联分析、拷贝数变异分析、Meta分析等等。
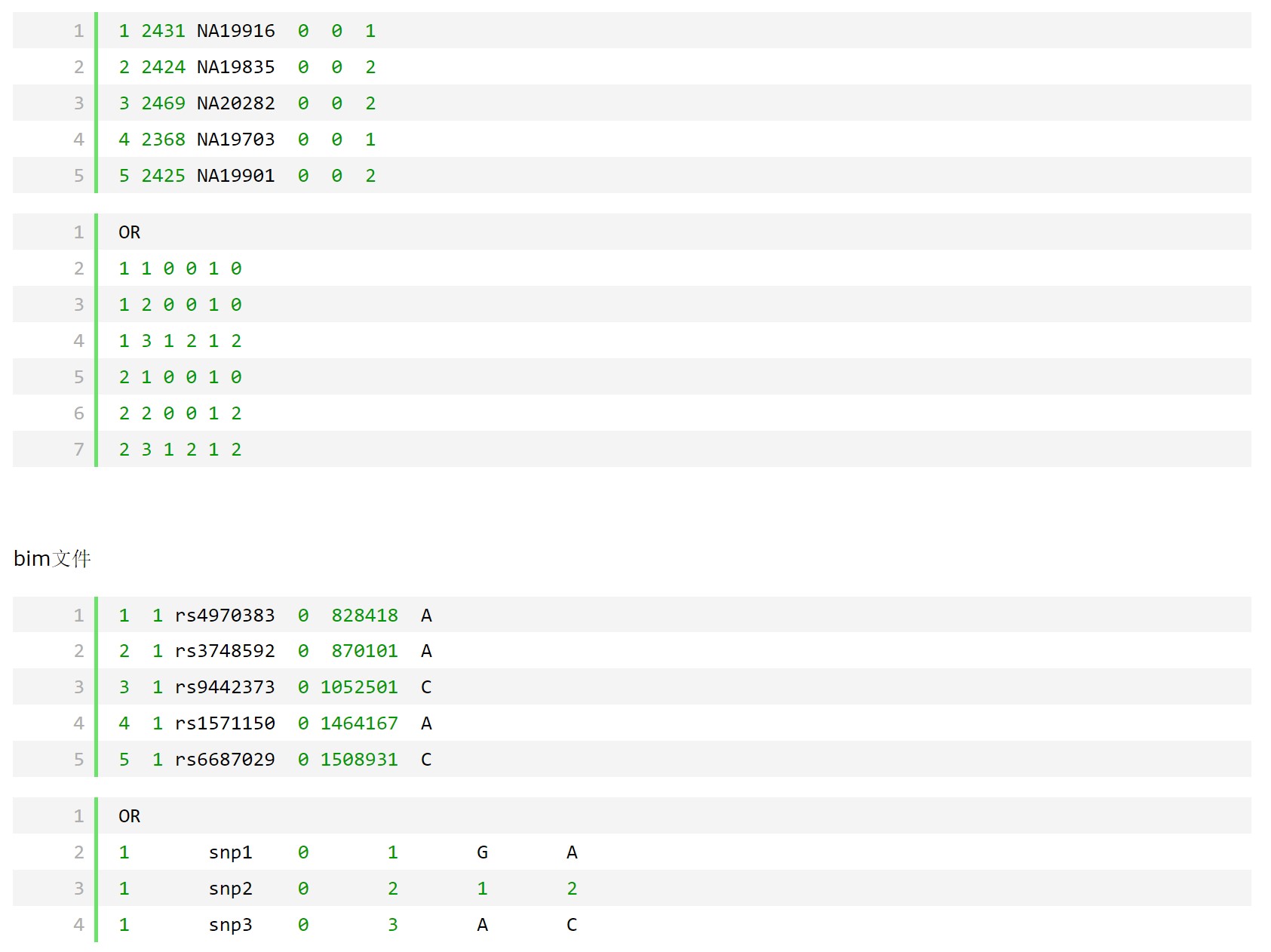
PLINK的三种格式:bed、fam和bim。(注意:这里的bed和genome里的区域文件bed完全不同)。
bed文件(真实的bed文件是二进制的,比较难读)
bed:Primary representation of genotype calls at biallelic variants. Must be accompanied by .bim and .fam files. Loaded with –bfile; generated in many situations, most notably when the –make-bed command is used. Do not confuse this with the UCSC Genome Browser’s BED format, which is totally different. 基因型信息。所以转换后就是一个matrix,每一行是一个个体,每一列就是一个变异。其中0、1、2分别对应了aa、Aa或aA和AA。不考虑碱基型,因为我们不关注ATGC的变化。
fam:Sample information file accompanying a .bed binary genotype table. 样本信息。每一行就是一个样本。
bim:Extended variant information file accompanying a .bed binary genotype table. 每一行是一个变异,及其注释信息。