参考资料:实践生物信息学
第一部分 使用Salmon对RNA-seq测序数据进行定量
1.1 整体流程
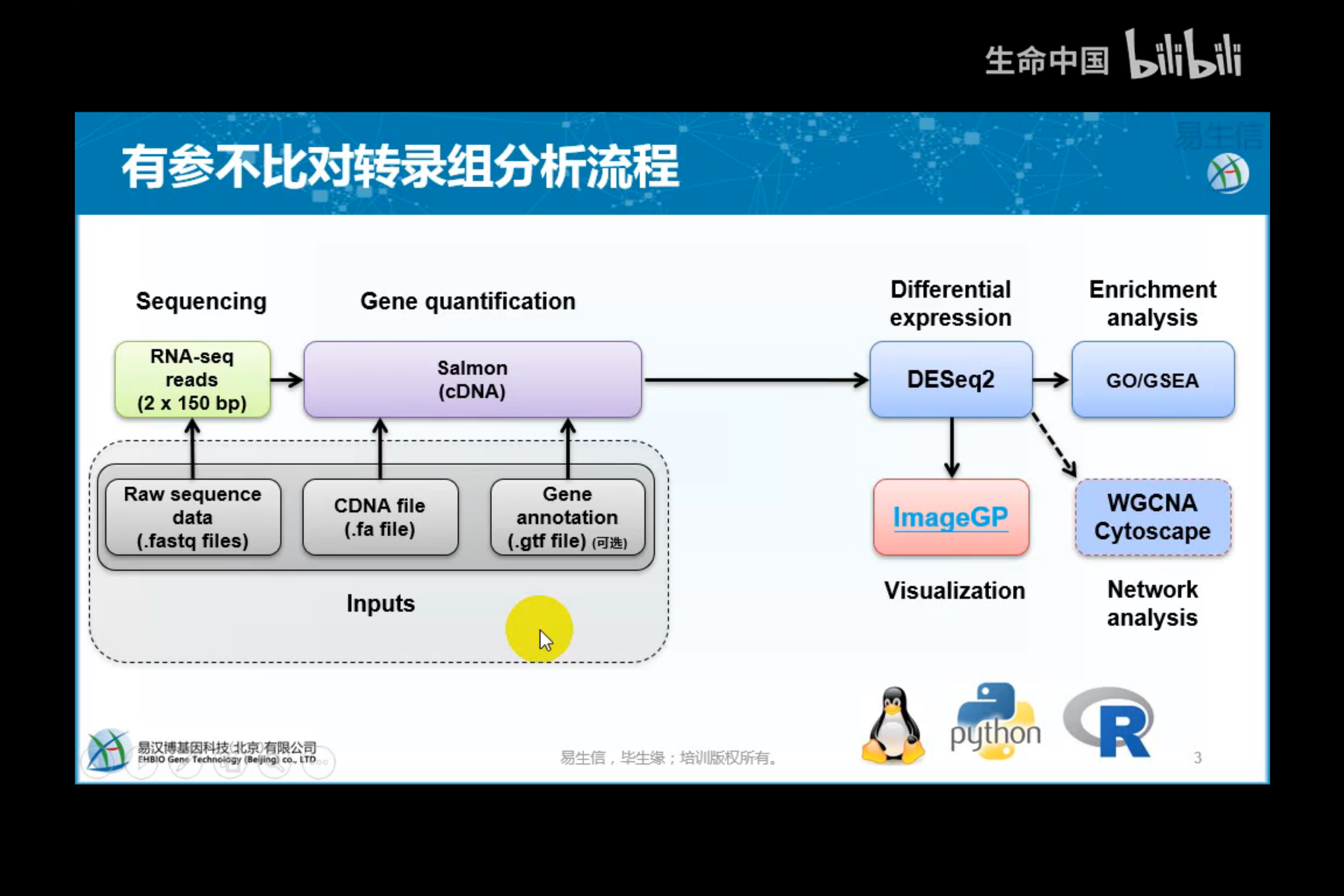
1.2 基因定量工具-Salmon的优势
-
定量时考虑到不同样品中基因长度的改变(比如不同isoform的使用)
-
速度快,需要的计算资源和存储资源小
-
敏感性高,不会丢弃匹配到多个基因同源区域的reads
-
可以直接校正GC-bias
-
自动判断文库类型
1.3 Salmon的输出结果

最主要的是下面两个:
- effective length
- TPM
cd ~/transcriptome/data
#从SRA文件中提取fastq文件
fastq-dump -v --split-3 --gzip SRR1039508
#测序质量评估
fastqc *.fq.gz
#multiqc整理评估结果
multiqc d . -o multiqc
#Salmon建立索引
salmon index -t GRCh38.transcript.fa -i GRCh38.salmon
#使用salmon进行定量
salmon quant --gcBias -l A -1 trt_N061011_1.fq.gz -2 trt_N061011_2.fq.gz -i genome/GRCh28.salmon -o trt_N061011/trt/N061011.salmon.count -p 4
#利用multiqc整理salmon的输出日志
multiqc -f -d . -o multiqc/
#列出salmon的输出文件,导出以用于后续DESeq2的分析
zip salmon.output.zip `find . -name quant.sf`
技巧1:结合SampleFile,用for循环代替手写命令进行批量定量**
for i in `tail -n +2 SampleFile | cut -f 1`
do
salmon quant -l A -1 ${i}_1.fq.gz -2 ${i}_2.fq.gz -i genome/GRCh38.salmon -o ${i}/${i}.salmon.count -p 4
done
第二部分 使用DESeq2对count结果进行差异分析
2.1 差异基因分析的几个问题
- 什么是差异基因分析?
不同组间稳定表达差异的基因筛选
- 为什么要做差异基因分析?
筛选靶点
- 怎么做差异基因分析?
标准化、统计差异检测、统计筛选
- 如果不进行标准化会怎样?
系统偏差
2.2 DESeq2进行差异分析的流程
输入文件为reads定量后的count_matrix文件:
setwd("/home/dingli/GSE81436/RNA-seq_Cr-DJ_osddm1a1b/DESeq_result")
countdata <- read.table("Cr-DJ_osddm1a1b_matrix.out", header=TRUE, row.names=1)
colnames(countdata) <- c("DJ-1","DJ-2","osddm1a1b-1","osddm1a1b-2")
condition <- factor(c("DJ","DJ","osddm1a1b","osddm1a1b"))
coldata <- data.frame(row.names=colnames(countdata), condition)
dds <- DESeqDataSetFromMatrix(countData=countdata, colData=coldata, design=~condition)
dds <- DESeq(dds)
res <- results(dds)
table(res$padj<0.01)
res_padj <- res[order(res$padj), ]
write.csv(res, file="diffexpr_padj_results.csv",row.names = T)
subset(res,padj < 0.01) -> diff
subset(diff,log2FoldChange < -1) -> down
subset(diff,log2FoldChange > 1) -> up
as.data.frame(down) -> down_gene
as.data.frame(up) -> up_gene
write.csv(up_gene, file="up_gene_LogFC1.csv",row.names = T)
write.csv(down_gene, file="down_gene_LogFC1.csv",row.names = T)