零、背景介绍
0.1 bed/bim/fam格式
0.1.1 bed文件
- bed:Primary representation of genotype calls at biallelic variants. (真实的bed文件是二进制的,比较难读)
- Must be accompanied by .bim and .fam files.
- Loaded with –bfile; generated in many situations, most notably when the –make-bed command is used.
- Do not confuse this with the UCSC Genome Browser’s BED format, which is totally different. (UCSC的bed文件是指基因型信息)。
- 所以转换后就是一个matrix,每一行是一个个体,每一列就是一个变异。其中0、1、2分别对应了aa、Aa或aA和AA。不考虑碱基型,因为我们不关注ATGC的变化。
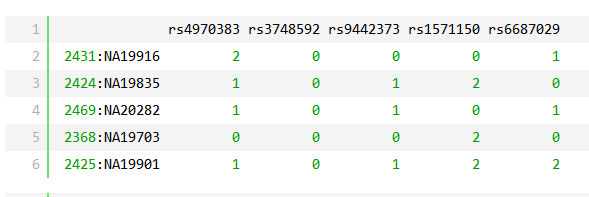
0.1.2 fam文件
- fam:Sample information file.每一行就是一个样本,共六列。
- 第一列:FID;
- 第二列:within-family ID;
- 第三列:within-family ID of father;
- 第四列:within-family ID of mother;
- 第五列:sex code (1 男 2 女 0 不知道);
- 第六列:Phenotype value。
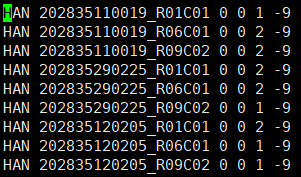
0.1.3 bim文件
- bim:Extended variant information file.
- 每一行是一个变异,及其注释信息。
- 第一列是染色体信息;
- 第二列是snp的名字;
- 第三列是摩尔距离,文件中说可以用0,没关系;
- 第四列是物理距离;
- 第五列是次要等位基因;
- 第六列是主要等位基因。
一、前序步骤:
1.1 用Genome Studio 进行Calling
Wegene直接给了二进制文件,所以不用这步。
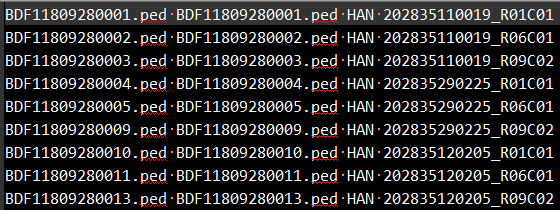
1.2 更新ids
Wegene给的二进制文件中,fam的IID和FID都是ped文件的名字,所以需要更新ids,改成HAN和ChipID,方便后续比较。文件形式如下,前两列是原FID和IID,后两列是需要更新的FID和IID。
plink –bfile data4914.uniq --update-ids renameIID.csv --noweb --make-bed -- new17
二、Individual QC
2.1 Sex QC
通过ChipID和金标准中的样本号做匹配,来提取性别信息,替换fam文件中的性别列(第五行)。可以通过字典匹配,注意不要打乱行的顺序。匹配完之后用下面命令运行:
plink --bfile data4914_clean_probe --check-sex --out sexstat --noweb
grep "PROBLEM" sexstat.sexcheck > error.sex
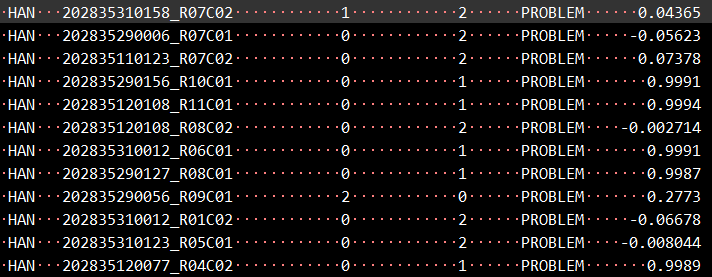
2.2 Missing rate QC
删除Missing rate>0.05的样本。
plink --bfile data4914_clean_probe --missing --out miss --noweb
awk '{$6>0.05}' miss.imiss > error.imiss
2.3 heterozygosity QC
删除杂合度高的样本,>0.2或<-0.2。
plink --bfile data4914_clean_probe --het --out het --noweb
awk '$6>=0.2 || $6<=-0.2' het.het > error.het

2.4 重复样本 QC
| 重复样本如果是样品放错则都删,如果知道是内参,则删missing rate高的样本。可以用cat miss.imiss | grep “202835290006_R07C01”查看missing rate。 |
plink --bfile data4914_clean_probe --indep-pairwise 50 5 0.2 --out ibd --noweb
plink --bfile data4914_clean_probe --extract ibd.prune.in --genome --out ibd --noweb
awk '$10>=0.95' ibd.genome > error.duplicate

2.5 合并需要删除的样本
将Sex中性别有误的人(性别缺失的不删,算不出来的和算错的删),可以参考如下命令,但记得性别那里要自己看一下哪些要删的:
cat error.indiv.* | sort -k 1 | uniq > error_indiv.txt,之后添加上"HAN"。
之后变成如下图所示的结果,放到一个文件中error_indiv.txt:
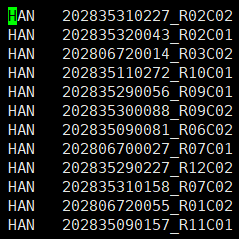
2.6 删除样本
通过上述的文件删除样本:
plink --bfile data4914_clean_probe --remove error_indiv.txt --make-bed --out clean_indiv --noweb
# 查看行数是否正确
wc -l clean_indiv.fam1
三、Marker QC
对位点进行QC。删除missing rate>0.02,HWE<0.00001,MAF<0.01的位点。
plink --bfile clean_indiv --geno 0.02 --hwe 0.00001 --maf 0.01 --make-bed --out clean_indiv_snp --noweb