1.安装并且加载scRNAseq
if (!requireNamespace("Rtsne"))
install.packages("Rtsne")
if (!requireNamespace("FactoMineR"))
install.packages("FactoMineR")
if (!requireNamespace("factoextra"))
install.packages("factoextra")
if (!requireNamespace("BiocManager"))
install.packages("BiocManager")
if (!requireNamespace("scater"))
BiocManager::install("scater")
if (!requireNamespace("scRNAseq"))
BiocManager::install("scRNAseq")
if (!requireNamespace("M3Drop"))
BiocManager::install("M3Drop")
if (!requireNamespace("ROCR"))
BiocManager::install("ROCR")
library(scater)
library(scRNAseq)
library(ggplot2)
library(tidyr)
library(cowplot)
library("FactoMineR")
library("factoextra")
library("ROCR")
2.scRNAseq R包中的数据集
这个包内置的是 Pollen et al. 2014 数据集,人类单细胞细胞,分成4类,分别是 pluripotent stem cells 分化而成的 neural progenitor cells (“NPC”) ,还有 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞,理解这些需要一定的生物学背景知识,如果不感兴趣,可以略过。
这个R包大小是50.6 MB,下载需要一点点时间,先安装加载它们。
这个数据集很出名,截止2019年1月已经有近400的引用了,后面的人开发R包算法都会在其上面做测试,比如 SinQC 这篇文章就提到:We applied SinQC to a highly heterogeneous scRNA-seq dataset containing 301 cells (mixture of 11 different cell types) (Pollen et al., 2014).
不过本例子只使用了数据集的4种细胞类型而已,因为 scRNAseq 这个R包就提供了这些,完整的数据是 23730 features, 301 samples, 地址为hemberg-lab, 这个网站非常值得推荐,简直是一个宝藏。
这里面的表达矩阵是由 RSEM (Li and Dewey 2011) 软件根据 hg38 RefSeq transcriptome 得到的,总是130个文库,每个细胞测了两次,测序深度不一样。
library(scRNAseq)
## ----- Load Example Data -----
fluidigm <- ReprocessedFluidigmData()
fluidigm
count <- floor(assays(fluidigm)$rsem_counts)
count[1:4,1:4]
## SRR1275356 SRR1274090 SRR1275251 SRR1275287
## A1BG 0 0 0 0
## A1BG-AS1 0 0 0 0
## A1CF 0 0 0 0
## A2M 0 0 0 33
sample_ann <- as.data.frame(colData(fluidigm))
DT::datatable(sample_ann)
3.先探索表型信息
前面说到,这个数据集是130个文库,每个细胞测了两次,测序深度不一样,这130个细胞,分成4类,分别是: pluripotent stem cells 分化而成的 neural progenitor cells (“NPC”) ,还有 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞。
批量,粗略的看一看各个细胞的一些统计学指标的分布情况
box <- lapply(colnames(sample_ann[,1:19]),function(i) {
dat <- sample_ann[,i,drop=F]
dat$sample=rownames(dat)
## 画boxplot
ggplot(dat, aes('all cells', get(i))) +
geom_boxplot() +
xlab(NULL)+ylab(i)
})
plot_grid(plotlist=box, ncol=5 )
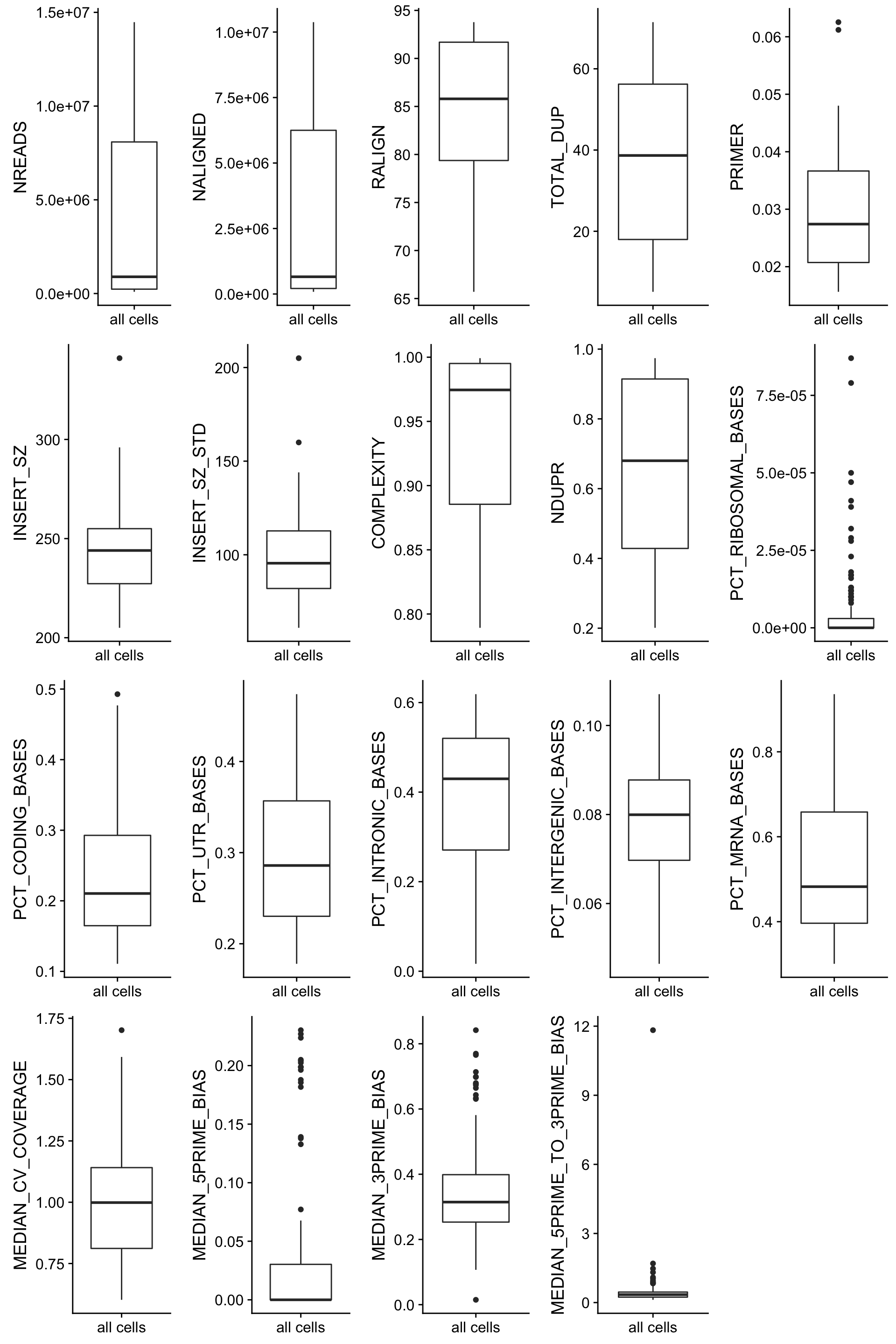
# ggsave(file="stat_all_cells.pdf")
很明显,他们可以有根据来进行分组,这里不再演示。 不过通常的文章并不会考虑如此多的细节,这里重点是批量,代码技巧非常值得你们学习。因为进行了简单探索,对表型数据就有了把握,接下来可以进行一定程度的过滤。
pa <- colnames(sample_ann[,c(1:9,11:16,18,19)])
tf <- lapply(pa,function(i) {
# i=pa[1]
dat <- sample_ann[,i]
dat <- abs(log10(dat))
fivenum(dat)
(up <- mean(dat)+2*sd(dat))
(down <- mean(dat)- 2*sd(dat) )
valid <- ifelse(dat > down & dat < up, 1,0 )
})
tf <- do.call(cbind,tf)
choosed_cells <- apply(tf,1,function(x) all(x==1))
table(sample_ann$Biological_Condition)
##
## GW16 GW21 GW21+3 NPC
## 52 16 32 30
sample_ann=sample_ann[choosed_cells,]
table(sample_ann$Biological_Condition)
##
## GW16 GW21 GW21+3 NPC
## 36 11 23 29
count[1:4,1:4]
## SRR1274090 SRR1275287 SRR1275364 SRR1275269
## A1BG 0 0 0 0
## A1BG-AS1 0 0 0 0
## A1CF 0 0 0 0
## A2M 0 33 0 51
fivenum(apply(counts,1,function(x) sum(x>0) ))
## A1CF OR8G1 LINC01003 MRPS36 YWHAZ
## 0 0 4 26 99
boxplot(apply(counts,1,function(x) sum(x>0) ))
fivenum(apply(counts,2,function(x) sum(x>0) ))
## SRR1275365 SRR1275345 SRR1275248 SRR1275273 SRR1274125
## 1566.0 3043.0 4002.0 5706.5 8024.0
hist(apply(counts,2,function(x) sum(x>0) ))
choosed_genes=apply(counts,1,function(x) sum(x>0) )>0
table(choosed_genes)
## choosed_genes
## FALSE TRUE
## 9496 16759
counts <- counts[choosed_genes,]
4.接下来要利用自己的常规转录组数据分析知识
4.1 看看细胞之间的所有的基因的表达量的相关性
下面的计算,都是基于log后的表达矩阵。
dat <- log2(edgeR::cpm(counts) + 1)
dat[1:4, 1:4]
## SRR1274090 SRR1275287 SRR1275364 SRR1275269
## A1BG 0 0.000000 0 0.000000
## A1BG-AS1 0 0.000000 0 0.000000
## A2M 0 4.216768 0 3.552694
## A2M-AS1 0 0.000000 0 0.000000
dat_back <- dat
先备份这个表达矩阵,后面的分析都用得上
exprSet <- dat_back
colnames(exprSet)
## [1] "SRR1274090" "SRR1275287" "SRR1275364" "SRR1275269" "SRR1275263"
## [6] "SRR1274117" "SRR1274089" "SRR1275248" "SRR1275345" "SRR1274125"
## [11] "SRR1275300" "SRR1275294" "SRR1274122" "SRR1275277" "SRR1275293"
## [16] "SRR1274128" "SRR1275348" "SRR1275245" "SRR1274084" "SRR1275342"
## [21] "SRR1275363" "SRR1275280" "SRR1275264" "SRR1275369" "SRR1274131"
## [26] "SRR1275351" "SRR1275256" "SRR1275355" "SRR1275258" "SRR1275260"
## [31] "SRR1275367" "SRR1275346" "SRR1274114" "SRR1275297" "SRR1275273"
## [36] "SRR1274126" "SRR1275290" "SRR1275274" "SRR1274121" "SRR1275341"
## [41] "SRR1275336" "SRR1274119" "SRR1274087" "SRR1274113" "SRR1275246"
## [46] "SRR1275267" "SRR1275289" "SRR1275360" "SRR1275358" "SRR1275352"
## [51] "SRR1274130" "SRR1275368" "SRR1275281" "SRR1275362" "SRR1275257"
## [56] "SRR1275350" "SRR1274129" "SRR1275298" "SRR1274123" "SRR1275343"
## [61] "SRR1275334" "SRR1274085" "SRR1274111" "SRR1275349" "SRR1275244"
## [66] "SRR1275344" "SRR1274088" "SRR1274116" "SRR1275243" "SRR1275301"
## [71] "SRR1275295" "SRR1275271" "SRR1274124" "SRR1275357" "SRR1275365"
## [76] "SRR1275268" "SRR1275361" "SRR1275266" "SRR1275288" "SRR1274133"
## [81] "SRR1275353" "SRR1275359" "SRR1274120" "SRR1275275" "SRR1275291"
## [86] "SRR1274086" "SRR1274112" "SRR1274118" "SRR1275340" "SRR1274115"
## [91] "SRR1275347" "SRR1274127" "SRR1275272" "SRR1275302" "SRR1275296"
## [96] "SRR1275354" "SRR1275259" "SRR1275285" "SRR1275366"
intall.packages("pheatmap")
library(pheatmap)
pheatmap::pheatmap(cor(exprSet))
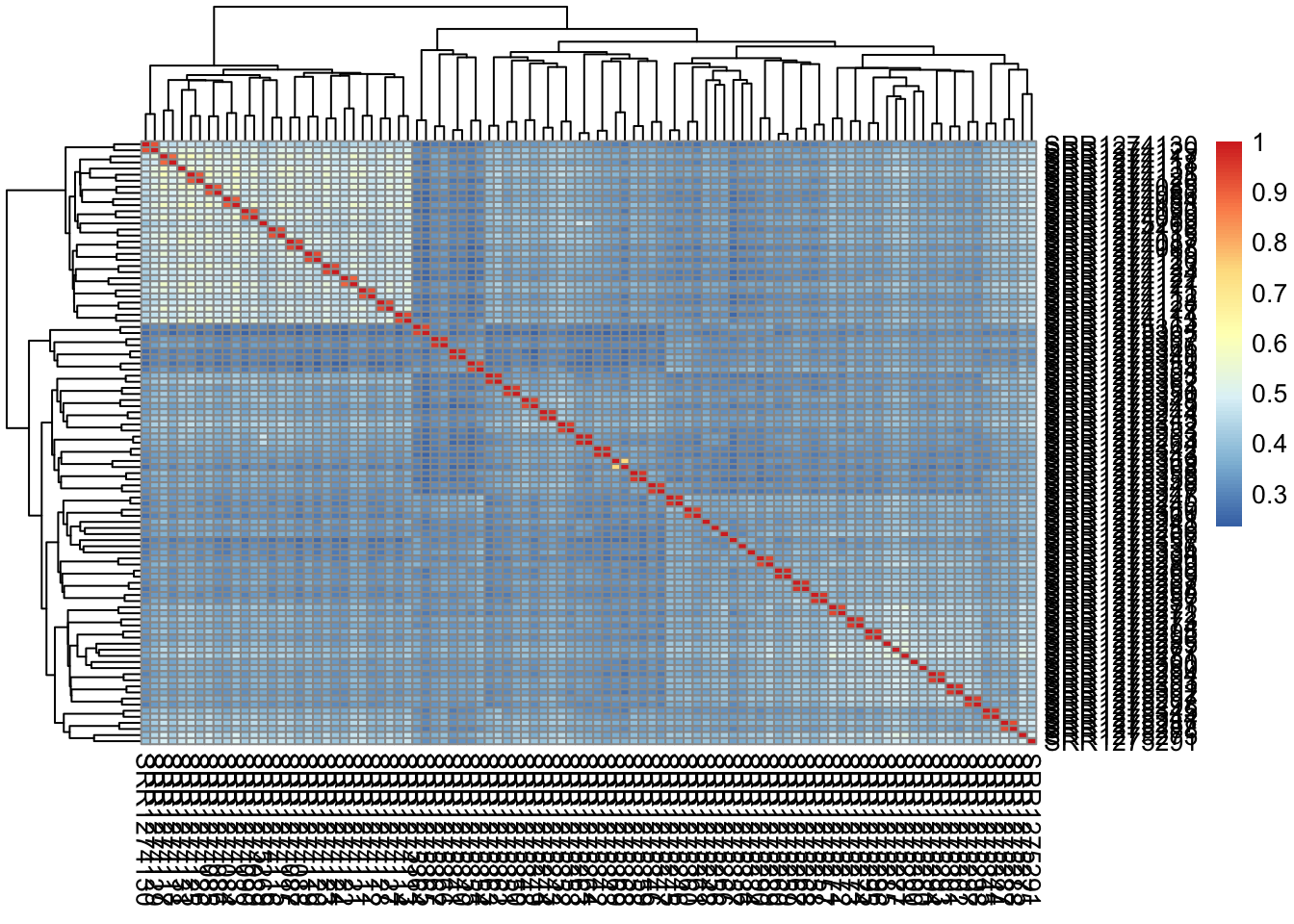
group_list <- sample_ann$Biological_Condition
tmp <- data.frame(g = group_list)
rownames(tmp) <- colnames(exprSet)
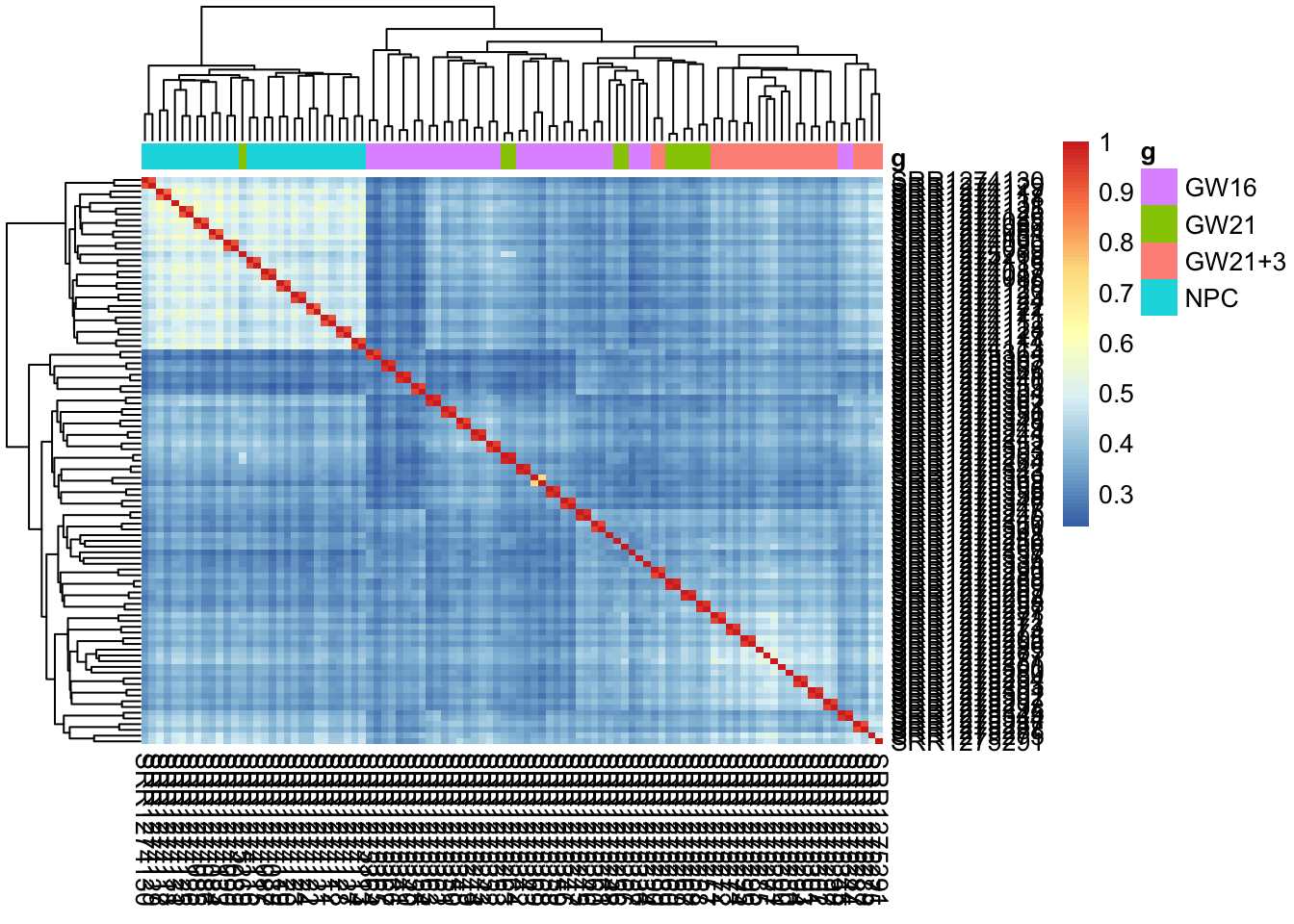
# 组内的样本的相似性应该是要高于组间的!
pheatmap::pheatmap(cor(exprSet), annotation_col = tmp)
dim(exprSet)
## [1] 16759 99
exprSet = exprSet[apply(exprSet, 1, function(x)
sum(x > 1) > 5), ]
dim(exprSet)
## [1] 11337 99
dim(exprSet)
## [1] 11337 99
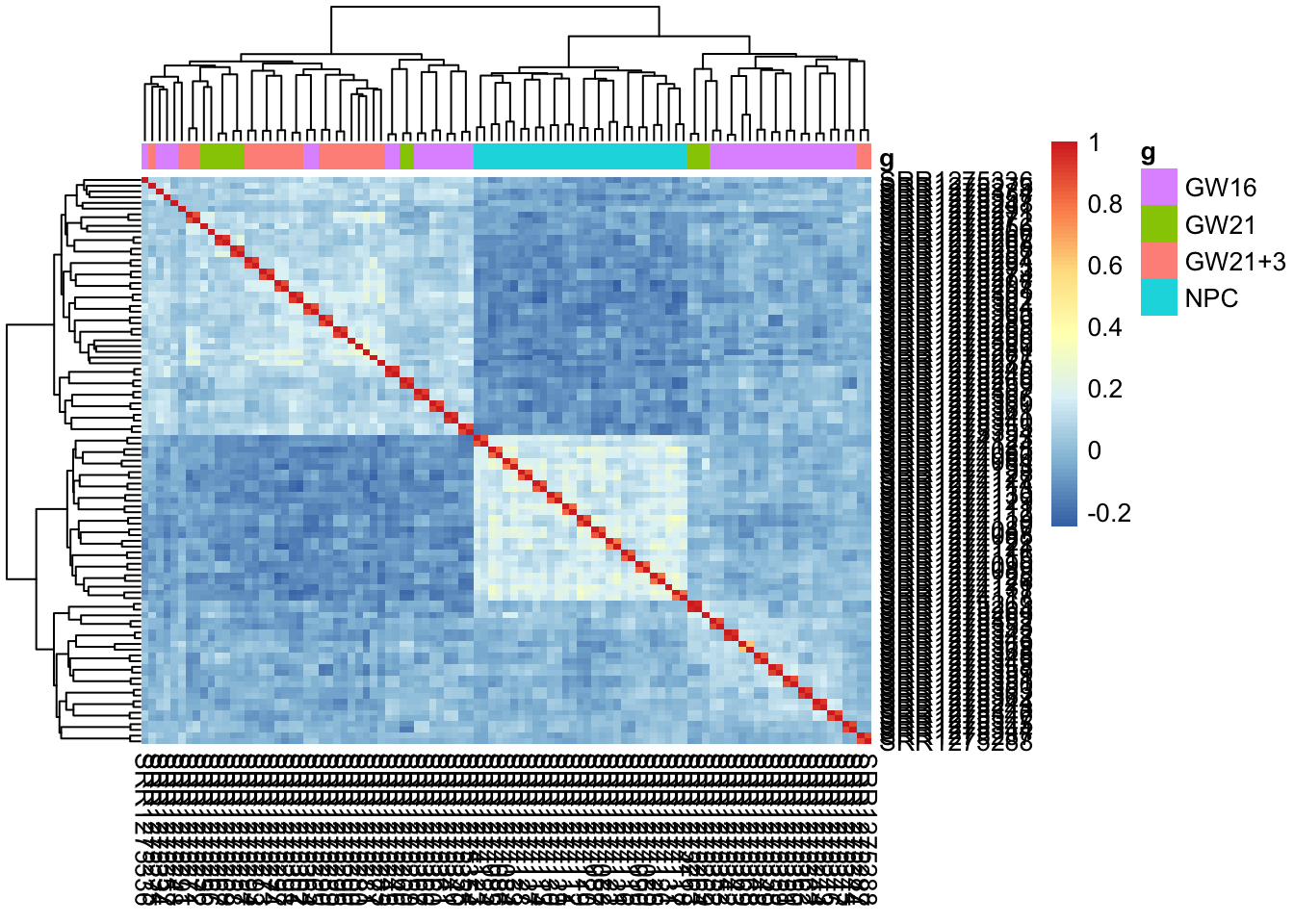
exprSet <- exprSet[names(sort(apply(exprSet, 1, mad), decreasing = T)[1:500]), ]
dim(exprSet)
## [1] 500 99
M <-cor(log2(exprSet + 1))
tmp <- data.frame(g = group_list)
rownames(tmp) <- colnames(M)
pheatmap::pheatmap(M, annotation_col = tmp)
table(sample_ann$LibraryName)
##
## GW16_1 GW16_10 GW16_12 GW16_13 GW16_14 GW16_15 GW16_16
## 1 1 2 2 2 2 2
## GW16_17 GW16_18 GW16_19 GW16_2 GW16_20 GW16_21 GW16_22
## 2 2 2 1 2 2 2
## GW16_23 GW16_24 GW16_25 GW16_4 GW16_5 GW16_6 GW21_1
## 2 2 2 2 2 1 1
## GW21_2 GW21_3 GW21_5 GW21_6 GW21_7 GW21_8 GW21+3_1
## 2 2 2 1 2 1 2
## GW21+3_10 GW21+3_11 GW21+3_12 GW21+3_13 GW21+3_14 GW21+3_16 GW21+3_2
## 2 1 1 1 1 1 2
## GW21+3_3 GW21+3_4 GW21+3_5 GW21+3_6 GW21+3_7 GW21+3_8 GW21+3_9
## 2 1 2 2 2 1 2
## NPC_1 NPC_10 NPC_11 NPC_12 NPC_13 NPC_14 NPC_15
## 2 2 2 2 2 2 2
## NPC_2 NPC_3 NPC_4 NPC_5 NPC_6 NPC_7 NPC_8
## 2 2 2 1 2 2 2
## NPC_9
## 2
可以看到,从细胞的相关性角度来看,到NPC跟另外的GW细胞群可以区分的很好,但是GW本身的3个小群体并没有那么好的区分度。
而且简单选取top的sd的基因来计算相关性,并没有很明显的改善。
但是可以看到每个细胞测了两次,所以它们的相关性要好于其它同类型的细胞。
4.2 对表达矩阵进行简单的层次聚类
如果计算机资源不够,这里可以先对基因进行一定程度的挑选,最简单的就是选取top的sd的基因,这里略。
dat <- dat_back
hc <- hclust(dist(t(dat)))
plot(hc,labels = FALSE)
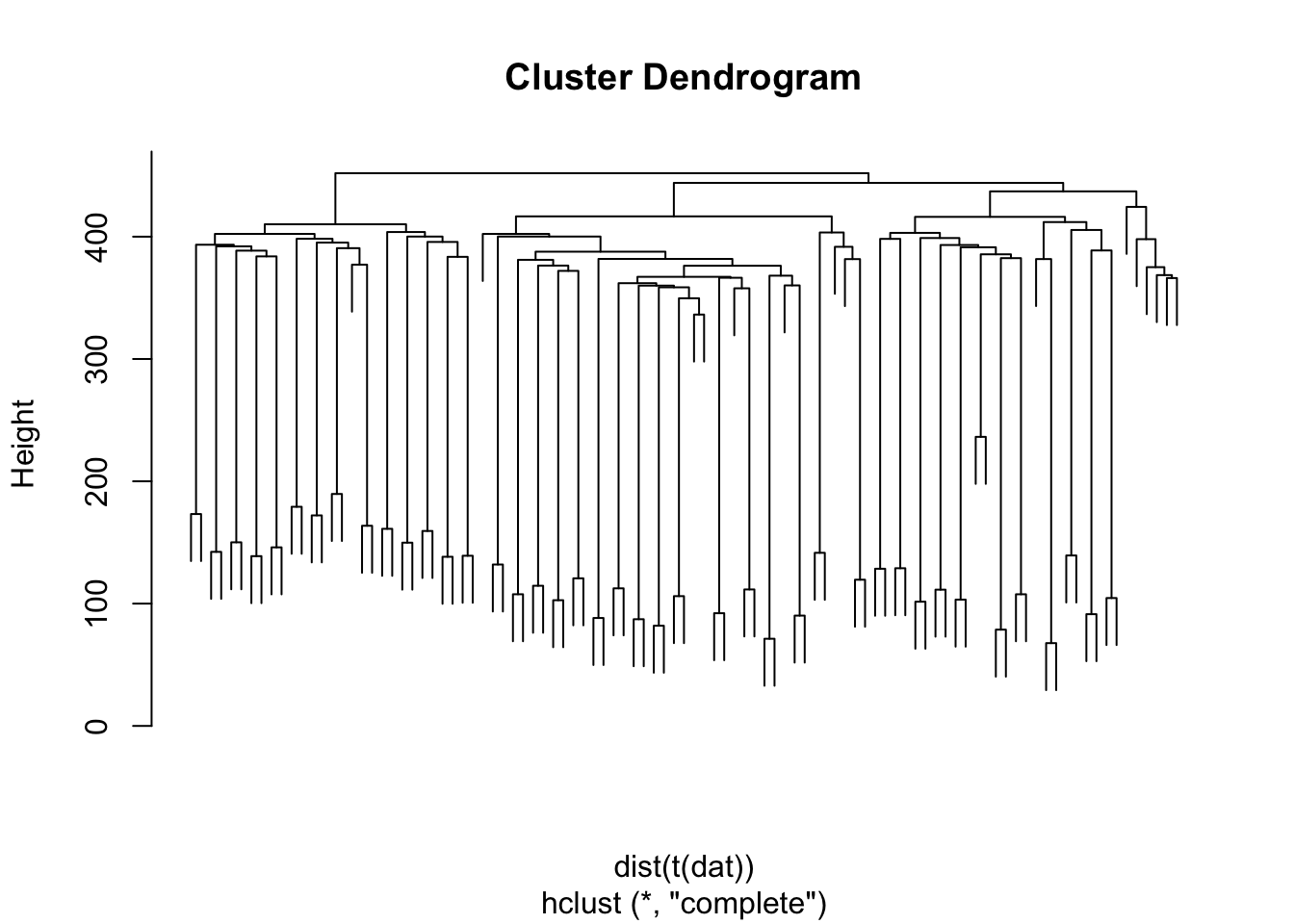
clus <- cutree(hc, 4) #对hclust()函数的聚类结果进行剪枝,即选择输出指定类别数的系谱聚类结果。
group_list <- as.factor(clus) ##转换为因子属性
table(group_list) ##统计频数
## group_list
## 1 2 3 4
## 29 25 39 6
table(group_list,sample_ann$Biological_Condition)
##
## group_list GW16 GW21 GW21+3 NPC
## 1 0 0 0 29
## 2 20 3 2 0
## 3 15 8 16 0
## 4 1 0 5 0
可以看到GW16和GW21是很难区分开来的,如果是普通的层次聚类的话。
4.3 看看最常规的PCA降维结果
降维算法很多,详情可以去自行搜索学习,比如:
-
主成分分析PCA
-
多维缩放(MDS)
-
线性判别分析(LDA)
-
等度量映射(Isomap)
-
局部线性嵌入(LLE)
-
t-SNE
-
Deep Autoencoder Networks
这里只介绍 PCA 和 t-SNE
dat <- dat_back
dat <- t(dat)
dat <- as.data.frame(dat)
plate <- sample_ann$Biological_Condition # 这里定义分组信息
dat <- cbind(dat, plate) #cbind根据列进行合并,即叠加所有列 #矩阵添加批次信息
dat[1:4, 1:4]
## A1BG A1BG-AS1 A2M A2M-AS1
## SRR1274090 0 0 0.000000 0
## SRR1275287 0 0 4.216768 0
## SRR1275364 0 0 0.000000 0
## SRR1275269 0 0 3.552694 0
table(dat$plate)
##
## GW16 GW21 GW21+3 NPC
## 36 11 23 29
# The variable plate (index = ) is removed
# before PCA analysis
dat.pca <- PCA(dat[, -ncol(dat)], graph = FALSE)
head(dat.pca$var$coord) ## 每个主成分的基因重要性占比
## Dim.1 Dim.2 Dim.3 Dim.4 Dim.5
## A1BG 0.19046450 0.09601240 -0.17840553 -0.001507970 -0.0006057691
## A1BG-AS1 -0.02510451 0.29821319 0.03571804 0.020001929 -0.0105727109
## A2M 0.03403042 0.25458727 0.24264958 0.228512329 0.5414019044
## A2M-AS1 0.23140893 0.02900348 -0.07952678 0.356461354 0.1283450099
## A2ML1 -0.15776536 0.13831288 0.10065788 0.004060288 -0.0353422367
## A2MP1 -0.04068586 -0.05584736 -0.02857416 0.018287992 0.0069603680
head(dat.pca$ind$coord) ## 每个细胞的前5个主成分取值。
## Dim.1 Dim.2 Dim.3 Dim.4 Dim.5
## SRR1274090 40.251912 -13.231641 -12.358891 -20.038100 -12.704947
## SRR1275287 1.196637 15.386256 30.566235 14.262858 -4.852418
## SRR1275364 -34.731051 -14.782146 -7.716928 7.046918 1.951473
## SRR1275269 21.760471 3.307309 17.985263 -18.382512 9.270646
## SRR1275263 -3.313968 -15.856721 8.929275 -36.358830 20.275875
## SRR1274117 59.378486 16.453551 -5.098901 56.245455 19.257598
fviz_pca_ind(
dat.pca,
#repel =T,
geom.ind = "point",
# show points only (nbut not "text")
col.ind = dat$plate,
# color by groups
#palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE,
# Concentration ellipses
legend.title = "Groups"
)
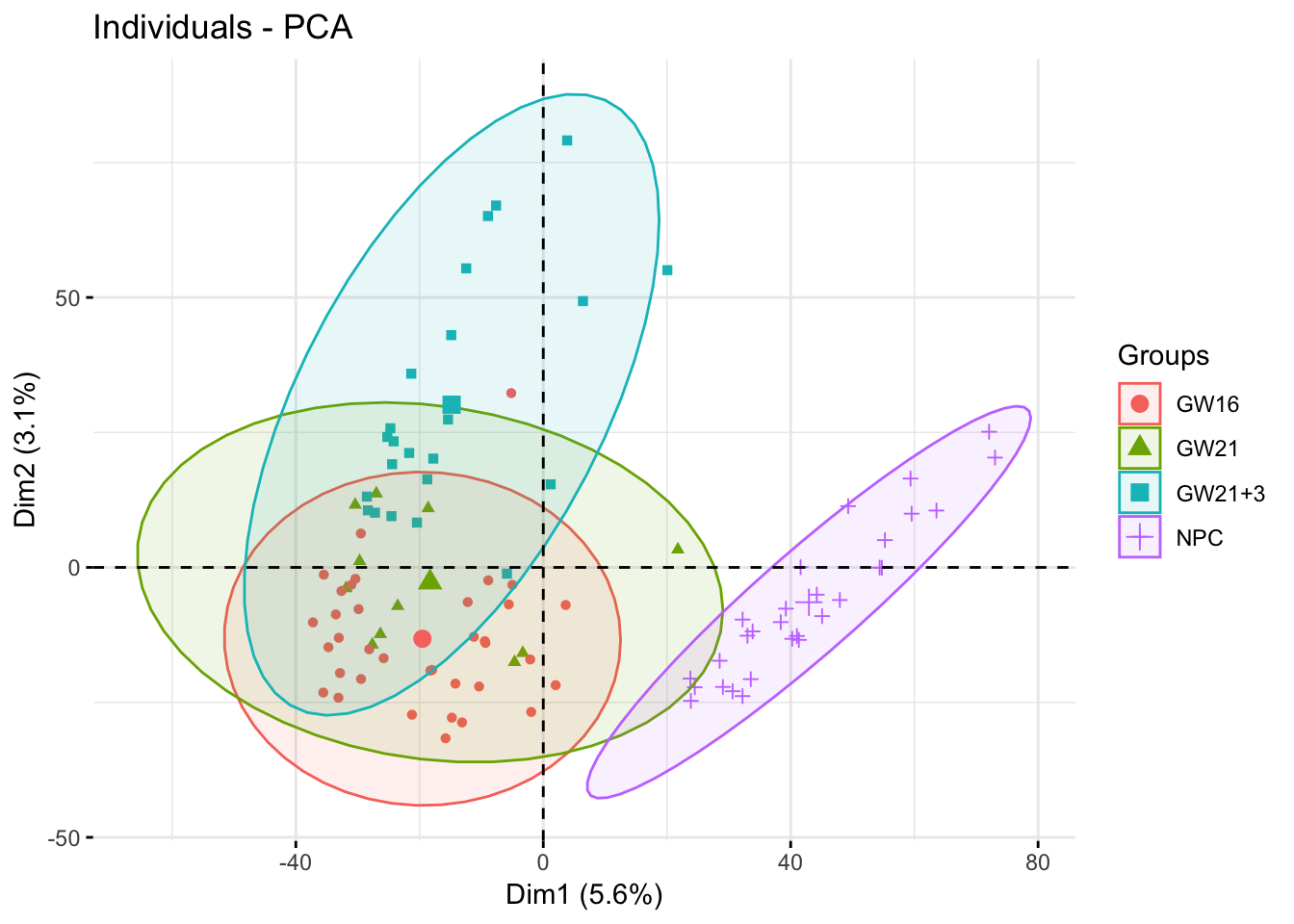
同样的,很明显可以看到NPC跟另外的GW细胞群可以区分的很好,但是GW本身的3个小群体并没有那么好的区分度。
4.4 接着是稍微高大上的tSNE降维
因为计算量的问题,这里先选取PCA后的主成分,然进行tSNE,当然,也有其它做法,比如选取变化高的基因,显著差异基因等等。
# 选取前面PCA分析的5个主成分。
dat_matrix <- dat.pca$ind$coord
# Set a seed if you want reproducible results
set.seed(42)
library(Rtsne)
# 如果使用原始表达矩阵进行 tSNE耗时很可怕,dat_matrix = dat_back
# 出现Remove duplicates before running TSNE 则check_duplicated = FALSE
# tsne_out <- Rtsne(dat_matrix,pca=FALSE,perplexity=30,theta=0.0, check_duplicates = FALSE) # Run TSNE
tsne_out <- Rtsne(dat_matrix,perplexity=10)
plate <- sample_ann$Biological_Condition # 这里定义分组信息
plot(tsne_out$Y,col= rainbow(4)[as.numeric(as.factor(plate))], pch=19)
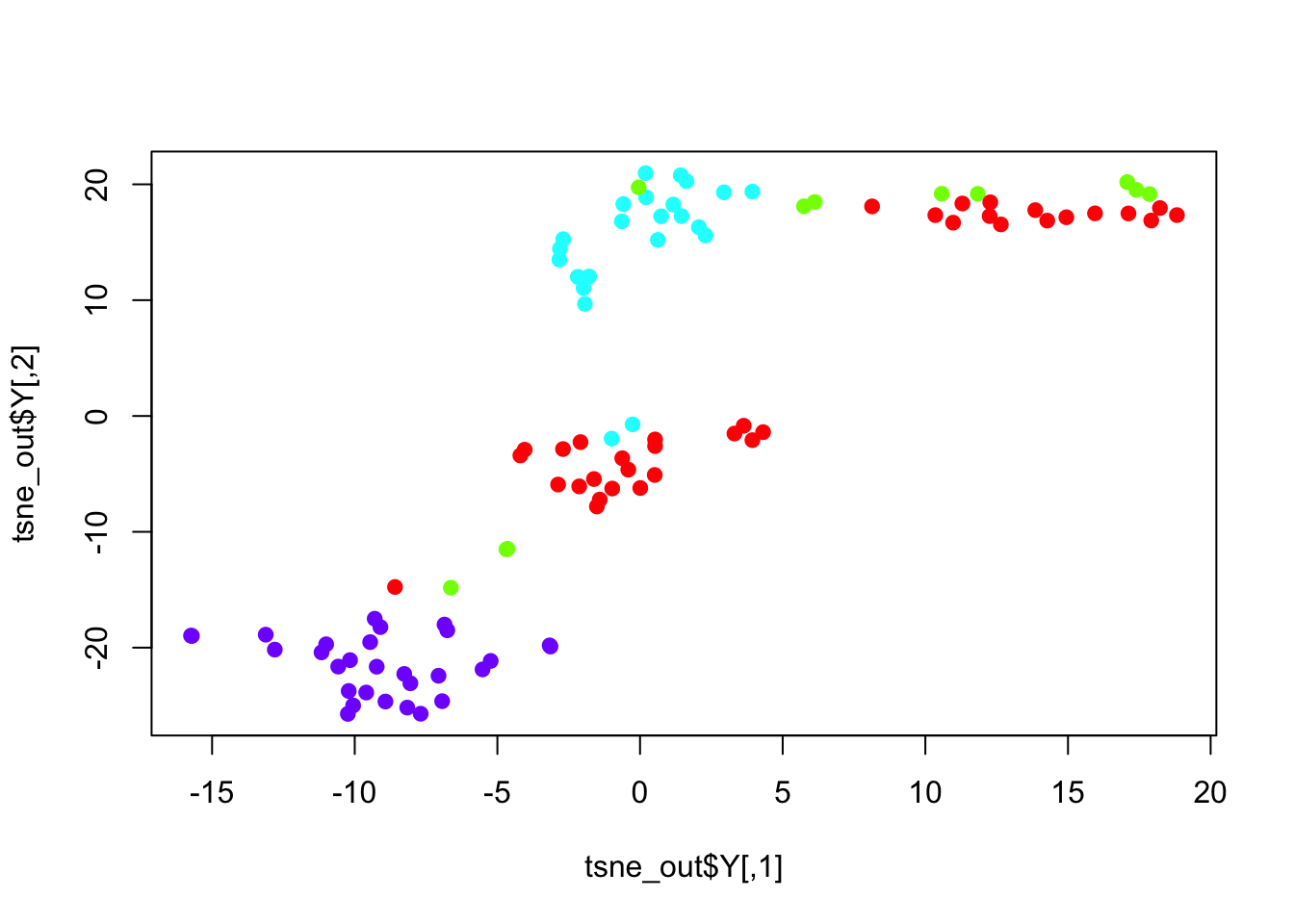
4.5 对PCA或者tSNE结果进行kmeans或者dbscan算法聚类
降维是降维,聚类是聚类,需要理解其中的区别。
降维与否,不同的降维算法选择,不同参数的选择得到的结果都不一样。
聚类也是一样,不同的算法,不同的参数。
# 前面我们的层次聚类是针对全部表达矩阵,这里我们为了节省计算量,可以使用tsne_out$Y这个结果
head(tsne_out$Y)
## [,1] [,2]
## [1,] -5.5201858 -21.8803070
## [2,] -0.2618134 -0.7269741
## [3,] 14.9423636 17.1572253
## [4,] -6.6313221 -14.8290355
## [5,] -4.6828620 -11.5136930
## [6,] -13.1192292 -18.8796564
opt_tsne=tsne_out$Y
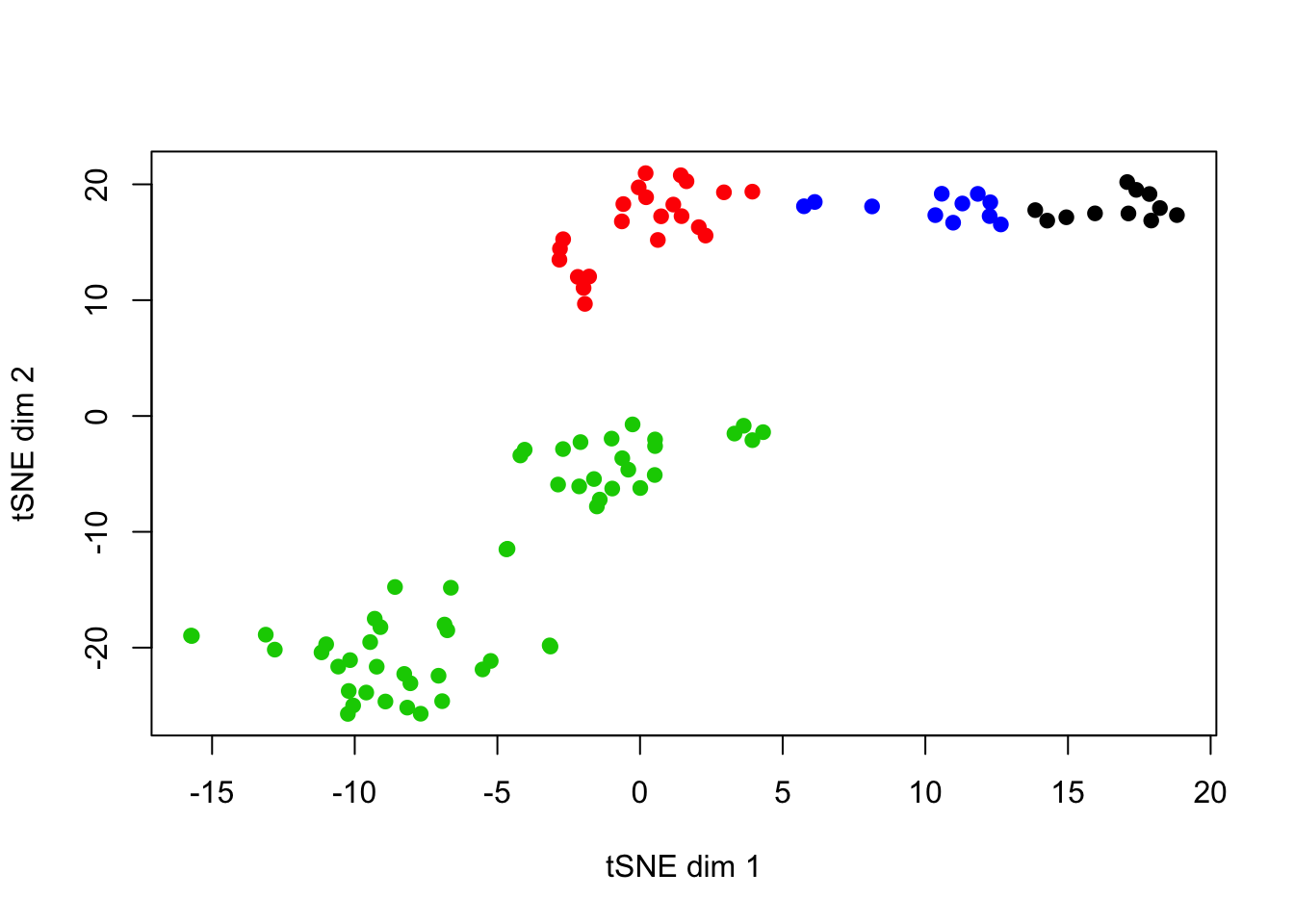
table(kmeans(opt_tsne,centers = 4)$clust)
##
## 1 2 3 4
## 22 15 18 44
plot(opt_tsne, col=kmeans(opt_tsne,centers = 4)$clust, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
library(dbscan)
plot(opt_tsne, col=dbscan(opt_tsne,eps=3.1)$cluster, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
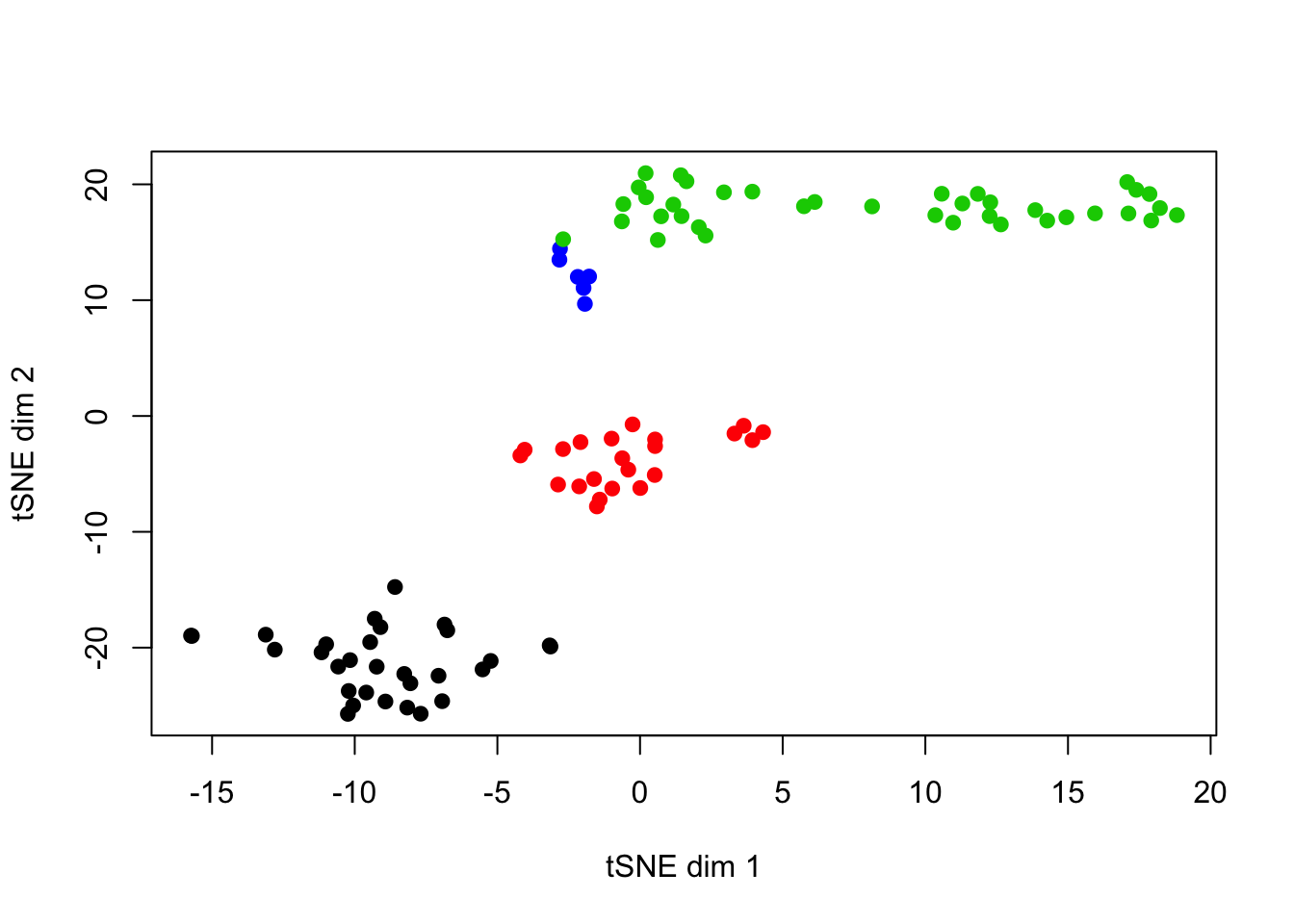
table(dbscan(opt_tsne,eps=3.1)$cluster)
##
## 0 1 2 3 4
## 3 30 22 38 6
# 比较两个聚类算法区别
table(kmeans(opt_tsne,centers = 4)$clust,dbscan(opt_tsne,eps=3.1)$cluster)
##
## 0 1 2 3 4
## 1 0 0 0 38 6
## 2 0 16 0 0 0
## 3 1 14 0 0 0
## 4 2 0 22 0 0
5.测试一下M3Drop这个单细胞转录组R包
5.1 首先构建M3Drop需要的对象
library(M3Drop)
Normalized_data <- M3DropCleanData(counts,
labels = sample_ann$Biological_Condition ,
is.counts=TRUE, min_detected_genes=2000)
dim(Normalized_data$data)
## [1] 13405 97
length(Normalized_data$labels)
## [1] 97
class(Normalized_data)
## [1] "list"
str(Normalized_data)
## List of 2
## $ data : num [1:13405, 1:97] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:13405] "A1BG" "A2M" "A2ML1" "AAAS" ...
## .. ..$ : chr [1:97] "SRR1274090" "SRR1275287" "SRR1275364" "SRR1275269" ...
## $ labels: chr [1:97] "NPC" "GW21+3" "GW16" "GW21" ...
这个包设计比较简单,并没有构建S4对象,只是一个简单的list而已。
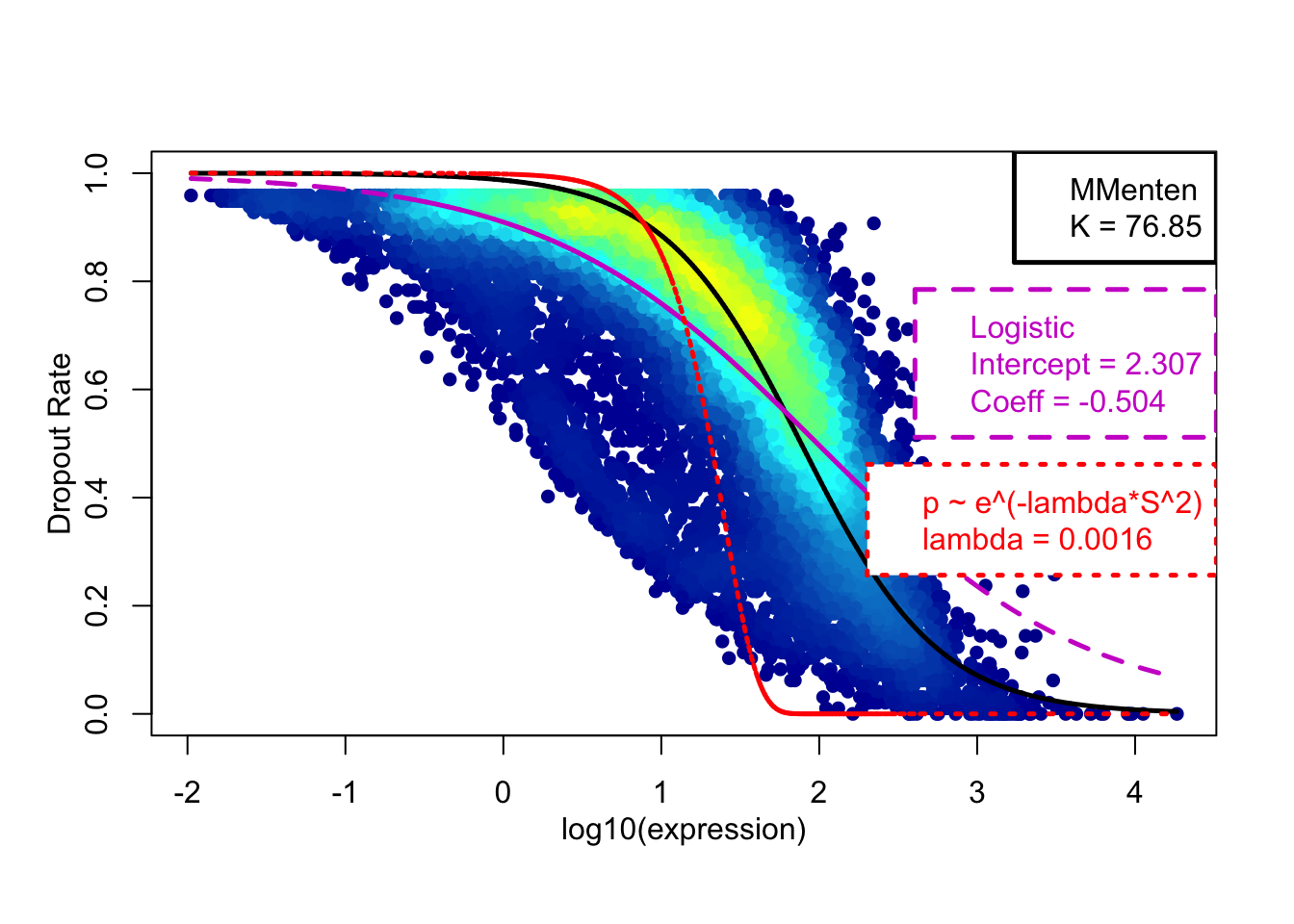
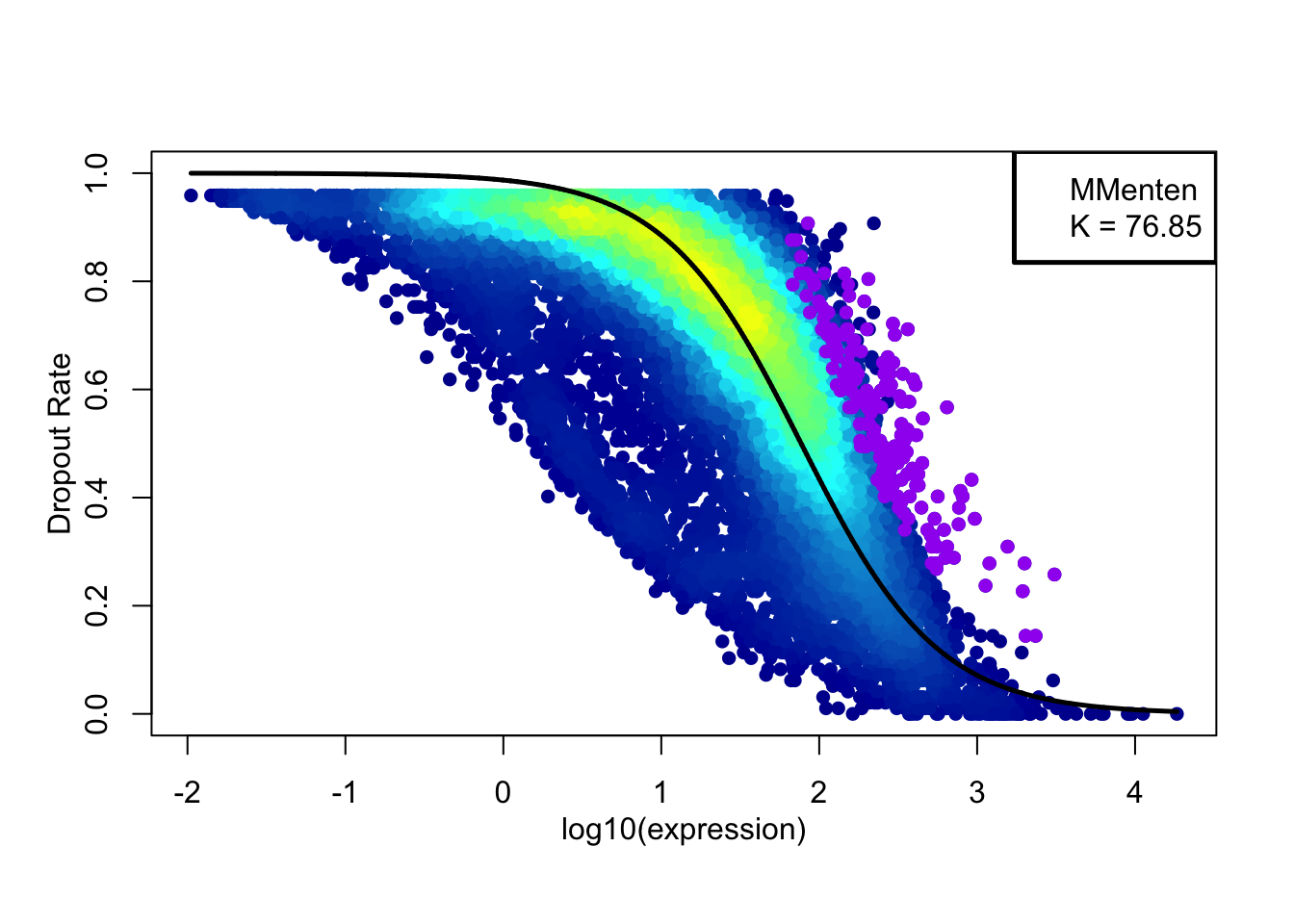
5.2 统计学算法 Michaelis-Menten
需要深入读该文章,了解其算法,这里略过,总之它对单细胞转录组的表达矩阵进行了一系列的统计检验。
fits <- M3DropDropoutModels(Normalized_data$data)
# Sum absolute residuals
data.frame(MM=fits$MMFit$SAr, Logistic=fits$LogiFit$SAr,
DoubleExpo=fits$ExpoFit$SAr)
## MM Logistic DoubleExpo
## 1 1651 1646 4033
# Sum squared residuals
data.frame(MM=fits$MMFit$SSr, Logistic=fits$LogiFit$SSr,
DoubleExpo=fits$ExpoFit$SSr)
## MM Logistic DoubleExpo
## 1 403 345 1962
5.3 找差异基因
DE_genes <- M3DropDifferentialExpression(Normalized_data$data,
mt_method="fdr", mt_threshold=0.01)
dim(DE_genes)
## [1] 180 3
head(DE_genes)
## Gene p.value q.value
## ABCE1 ABCE1 2.844523e-05 3.020909e-03
## ACAT2 ACAT2 7.885891e-05 6.776306e-03
## ADGRB3 ADGRB3 1.083437e-04 8.443879e-03
## AMER2 AMER2 1.917979e-05 2.255308e-03
## ANKRD44 ANKRD44 6.749811e-05 6.113596e-03
## ANP32E ANP32E 1.547604e-07 3.771932e-05
这里是针对上面的统计结果来的
5.4 针对差异基因画热图
par(mar=c(1,1,1,1))
heat_out <- M3DropExpressionHeatmap(DE_genes$Gene, Normalized_data$data,
cell_labels = Normalized_data$labels)
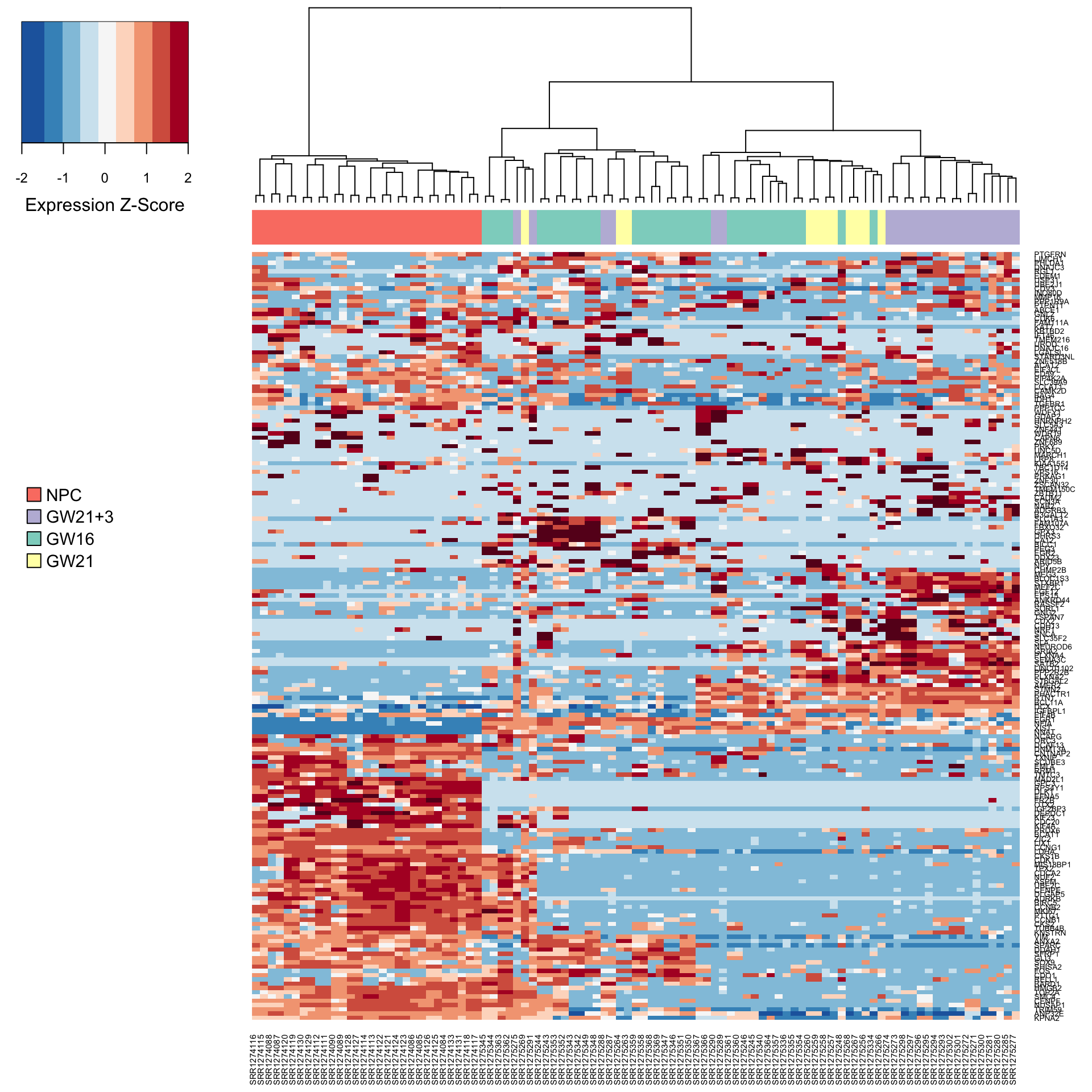
可视化了解一下找到的差异基因在不同的细胞类型的表达分布情况。
5.4 聚类
这里可以重新聚类后,针对自己找到的类别来分别找marker基因,不需要使用测试数据自带的表型信息。
cell_populations <- M3DropGetHeatmapCellClusters(heat_out, k=4)
library("ROCR")
marker_genes <- M3DropGetMarkers(Normalized_data$data, cell_populations)
table(cell_populations,Normalized_data$labels)
##
## cell_populations GW16 GW21 GW21+3 NPC
## 1 0 0 0 29
## 2 14 8 19 0
## 3 4 1 2 0
## 4 16 2 2 0
5.5 每个类别的marker genes
head(marker_genes[marker_genes$Group==4,],20)
## AUC Group pval
## ADGRV1 0.9707792 4 1.831217e-11
## TFAP2C 0.9451299 4 1.885637e-11
## EGR1 0.9409091 4 1.159852e-09
## PLCE1 0.9233766 4 7.676760e-11
## FOS 0.9058442 4 1.806229e-08
## SLC1A3 0.9048701 4 1.542660e-09
## AASS 0.9032468 4 3.136961e-08
## ITGB8 0.8886364 4 8.823909e-08
## BCAN 0.8844156 4 1.513264e-12
## NFIA 0.8831169 4 7.724889e-08
## PTPRZ1 0.8707792 4 3.596940e-07
## HES1 0.8590909 4 7.233415e-08
## DUSP10 0.8584416 4 7.865906e-09
## LTBP1 0.8545455 4 1.872381e-07
## LDLR 0.8487013 4 1.547451e-06
## FBXO32 0.8477273 4 3.810723e-09
## CLU 0.8467532 4 1.319982e-06
## CCND2 0.8422078 4 2.668888e-06
## JUN 0.8357143 4 8.979968e-07
## GPM6B 0.8350649 4 4.121035e-06
marker_genes[rownames(marker_genes)=="FOS",]
## AUC Group pval
## FOS 0.9058442 4 1.806229e-08
也可以针对这些 marker genes去画热图,当然,得根据AUC和P值来挑选符合要求的差异基因去绘图。
par(mar=c(1,1,1,1))
choosed_marker_genes=as.character(unlist(lapply(split(marker_genes,marker_genes$Group), function(x) (rownames(head(x,20))))))
heat_out <- M3DropExpressionHeatmap(choosed_marker_genes, Normalized_data$data, cell_labels = cell_populations)
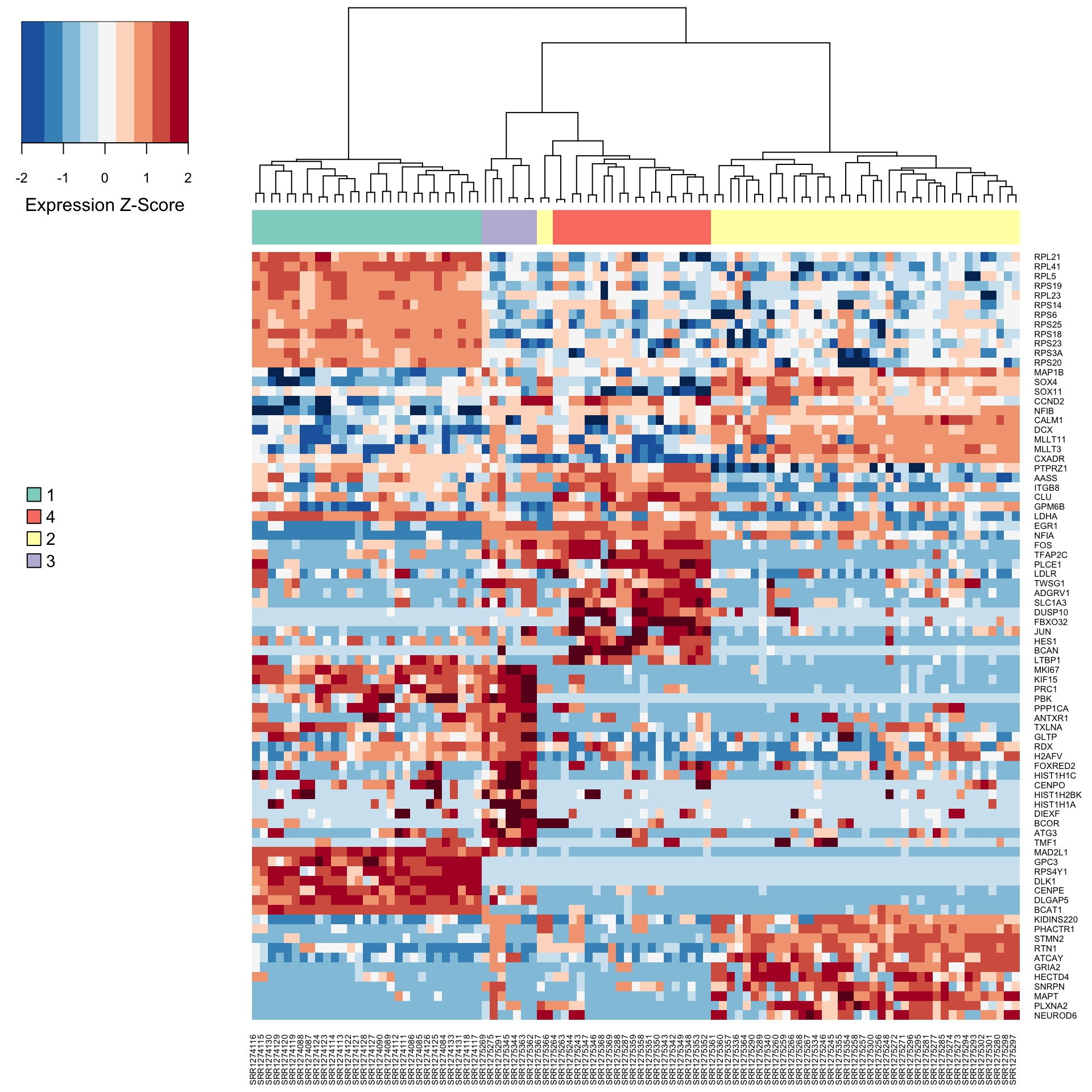
如果遇到Error in plot.new() : figure margins too large报错,则单独将heat_out这行命令复制出来运行
6.对感兴趣基因集进行注释
通常是GO/KEGG等数据库,通常是超几何分布,GSEA,GSVA等算法。
这里就略过。