0.引言
教程,当然是以官网为主,不过看英文笔记有挑战,简略带领大家一起学习咯: Biconductor-Scater
值得提醒的是 2017年 11 月 这个 scater 包经过了重大变革,所以如果大家看到比较旧的教程需要注意一下,通常是无法成功的。
其GitHub的教程:hemberg-lab
1.载入必要的R包
需要自行下载安装一些必要的R包! 因为大量学员在中国大陆,通常不建议大家使用下面的R包安装方法,建议是切换镜像后再下载R包。参考:生信技能树
if (!requireNamespace("Rtsne"))
install.packages("Rtsne")
if (!requireNamespace("BiocManager"))
install.packages("BiocManager")
if (!requireNamespace("scater"))
BiocManager::install("scater")
if (!requireNamespace("scRNAseq"))
BiocManager::install("scRNAseq")
if (!requireNamespace("destiny"))
BiocManager::install("destiny")
if (!requireNamespace("SC3"))
BiocManager::install("SC3")
加载R包
rm(list = ls()) # clear the environment
#load all the necessary libraries
options(warn=-1) # turn off warning message globally
suppressMessages(library(scater))
suppressMessages(library(scRNAseq))
2.创建测试数据集
我们选择 scRNAseq 这个R包。 这个包内置的是 Pollen et al. 2014 数据集,人类单细胞细胞,分成4类,分别是 pluripotent stem cells 分化而成的 neural progenitor cells (“NPC”) ,还有 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞。大小是50.6 MB,下载需要一点点时间,先安装加载它们。
这个数据集很出名,截止2019年1月已经有近400的引用了,后面的人开发R包算法都会在其上面做测试,比如 SinQC 这篇文章就提到:We applied SinQC to a highly heterogeneous scRNA-seq dataset containing 301 cells (mixture of 11 different cell types) (Pollen et al., 2014).
不过本例子只使用了数据集的4种细胞类型而已,因为 scRNAseq 这个R包就提供了这些,完整的数据是 23730 features, 301 samples 在 https://hemberg-lab.github.io/scRNA.seq.datasets/human/tissues/
这里面的表达矩阵是由 RSEM (Li and Dewey 2011) 软件根据 hg38 RefSeq transcriptome 得到的,总是130个文库,每个细胞测了两次,测序深度不一样。
library(scRNAseq)
## ----- Load Example Data -----
fluidigm <- ReprocessedFluidigmData()
fluidigm
# Set assay to RSEM estimated counts
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
## SRR1275356 SRR1274090 SRR1275251 SRR1275287
## A1BG 0 0 0 0
## A1BG-AS1 0 0 0 0
## A1CF 0 0 0 0
## A2M 0 0 0 33
table(rowSums(ct)==0)
##
## FALSE TRUE
## 17096 9159
# 这里使用原始表达矩阵,所以有很多基因在所有细胞均无表达量,即表现为没有被检测到,这样的基因是需要过滤掉的。
pheno_data <- as.data.frame(colData(fluidigm))
## 这里需要把Pollen的表达矩阵做成我们的 scater 要求的对象
#data("sc_example_counts")
#data("sc_example_cell_info")
# 你也可以尝试该R包自带的数据集。
# 参考 https://bioconductor.org/packages/release/bioc/vignettes/scater/inst/doc/vignette-intro.R
sce <- SingleCellExperiment(
assays = list(counts = ct),
colData = pheno_data
)
sce
## class: SingleCellExperiment
## dim: 26255 130
## metadata(0):
## assays(1): counts
## rownames(26255): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(0):
## colnames(130): SRR1275356 SRR1274090 ... SRR1275366 SRR1275261
## colData names(28): NREADS NALIGNED ... Cluster1 Cluster2
## reducedDimNames(0):
## spikeNames(0):
# 后面所有的分析都是基于 sce 这个变量
# 是一个 SingleCellExperiment 对象,被很多单细胞R包采用。
3.一些质量控制
这个也非常复杂,推荐看原文说明书: https://www.bioconductor.org/packages/devel/bioc/vignettes/scater/inst/doc/vignette-qc.html
这里稍作讲解,包括基因层面及细胞层面,但是并不设计实验本身引入的干扰因素。
3.1 首先看基因层面的过滤
使用 calculateQCMetrics 函数作用于 sce 那个单细胞数据对象后,就可以 用 rowData(object) 可以查看各个基因统计指标:
- mean_counts: 平均表达量
- log10_mean_counts: 归一化 log10-scale.
- pct_dropout_by_counts: 该基因丢失率。
- n_cells_by_counts: 多少个细胞表达了该基因
上面那些指标可以用来过滤,也可以自己重新再次计算一下那些统计学指标。
主要是过滤低表达量基因,还有 线粒体基因 和 ERCC spike-ins 的控制。
exprs(sce) <- log2(
calculateCPM(sce ) + 1)
## 只有运行了下面的函数后才有各式各样的过滤指标
genes <- rownames(rowData(sce))
genes[grepl('^MT-',genes)]
## character(0)
genes[grepl('^ERCC-',genes)]
## character(0)
# 比较不幸的是,这个测试数据里面既没有线粒体基因,又没有ERCC序列。
sce <- calculateQCMetrics(sce,
feature_controls = list(ERCC = grep('^ERCC',genes)))
# 也没有定义啥细胞是属于control组,所以这里并不需要完全follow教程
# example_sce <- perCellQCMetrics(example_sce,
# feature_controls = list(ERCC = 1:20, mito = 500:1000),
# cell_controls = list(empty = 1:5, damaged = 31:40))
查看信息
tmp <- as.data.frame(rowData(sce))
colnames(tmp)
## [1] "is_feature_control" "is_feature_control_ERCC"
## [3] "mean_counts" "log10_mean_counts"
## [5] "n_cells_by_counts" "pct_dropout_by_counts"
## [7] "total_counts" "log10_total_counts"
head(tmp)
## is_feature_control is_feature_control_ERCC mean_counts
## A1BG FALSE FALSE 1.553846e+00
## A1BG-AS1 FALSE FALSE 1.015385e+00
## A1CF FALSE FALSE 7.692308e-03
## A2M FALSE FALSE 1.505154e+02
## A2M-AS1 FALSE FALSE 1.407692e+00
## A2ML1 FALSE FALSE 1.538462e-01
## log10_mean_counts n_cells_by_counts pct_dropout_by_counts
## A1BG 0.407194731 10 92.30769
## A1BG-AS1 0.304357939 2 98.46154
## A1CF 0.003327943 1 99.23077
## A2M 2.180456733 21 83.84615
## A2M-AS1 0.381600985 3 97.69231
## A2ML1 0.062147907 9 93.07692
## total_counts log10_total_counts
## A1BG 202 2.307496
## A1BG-AS1 132 2.123852
## A1CF 1 0.301030
## A2M 19567 4.291546
## A2M-AS1 183 2.264818
## A2ML1 20 1.322219
目前只过滤那些在所有细胞都没有表达的基因, 但是这个过滤条件可以自行调整。可以看到基因数量大幅度减少。
keep_feature <- rowSums(exprs(sce) > 0) > 0
table(keep_feature)
## keep_feature
## FALSE TRUE
## 9159 17096
sce <- sce[keep_feature,]
sce
## class: SingleCellExperiment
## dim: 17096 130
## metadata(0):
## assays(2): counts logcounts
## rownames(17096): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(8): is_feature_control is_feature_control_ERCC ...
## total_counts log10_total_counts
## colnames(130): SRR1275356 SRR1274090 ... SRR1275366 SRR1275261
## colData names(64): NREADS NALIGNED ...
## pct_counts_in_top_200_features_ERCC
## pct_counts_in_top_500_features_ERCC
## reducedDimNames(0):
## spikeNames(0):
3.2 然后看细胞层面的过滤
用 colData(object) 可以查看各个样本统计情况
-
total_counts: total number of counts for the cell (aka ‘library size’)
-
log10_total_counts: total_counts on the log10-scale
-
total_features: the number of features for the cell that have expression above the detection limit (default detection limit is zero)
等等
每个数据集都有适合自己的过滤阈值,不要局限于教程
tmp <- as.data.frame(colData(sce))
colnames(tmp)
## [1] "NREADS"
## [2] "NALIGNED"
## [3] "RALIGN"
## [4] "TOTAL_DUP"
## [5] "PRIMER"
## [6] "INSERT_SZ"
## [7] "INSERT_SZ_STD"
## [8] "COMPLEXITY"
## [9] "NDUPR"
## [10] "PCT_RIBOSOMAL_BASES"
## [11] "PCT_CODING_BASES"
## [12] "PCT_UTR_BASES"
## [13] "PCT_INTRONIC_BASES"
## [14] "PCT_INTERGENIC_BASES"
## [15] "PCT_MRNA_BASES"
## [16] "MEDIAN_CV_COVERAGE"
## [17] "MEDIAN_5PRIME_BIAS"
## [18] "MEDIAN_3PRIME_BIAS"
## [19] "MEDIAN_5PRIME_TO_3PRIME_BIAS"
## [20] "sample_id.x"
## [21] "Lane_ID"
## [22] "LibraryName"
## [23] "avgLength"
## [24] "spots"
## [25] "Biological_Condition"
## [26] "Coverage_Type"
## [27] "Cluster1"
## [28] "Cluster2"
## [29] "is_cell_control"
## [30] "total_features_by_counts"
## [31] "log10_total_features_by_counts"
## [32] "total_counts"
## [33] "log10_total_counts"
## [34] "pct_counts_in_top_50_features"
## [35] "pct_counts_in_top_100_features"
## [36] "pct_counts_in_top_200_features"
## [37] "pct_counts_in_top_500_features"
## [38] "total_features_by_counts_endogenous"
## [39] "log10_total_features_by_counts_endogenous"
## [40] "total_counts_endogenous"
## [41] "log10_total_counts_endogenous"
## [42] "pct_counts_endogenous"
## [43] "pct_counts_in_top_50_features_endogenous"
## [44] "pct_counts_in_top_100_features_endogenous"
## [45] "pct_counts_in_top_200_features_endogenous"
## [46] "pct_counts_in_top_500_features_endogenous"
## [47] "total_features_by_counts_feature_control"
## [48] "log10_total_features_by_counts_feature_control"
## [49] "total_counts_feature_control"
## [50] "log10_total_counts_feature_control"
## [51] "pct_counts_feature_control"
## [52] "pct_counts_in_top_50_features_feature_control"
## [53] "pct_counts_in_top_100_features_feature_control"
## [54] "pct_counts_in_top_200_features_feature_control"
## [55] "pct_counts_in_top_500_features_feature_control"
## [56] "total_features_by_counts_ERCC"
## [57] "log10_total_features_by_counts_ERCC"
## [58] "total_counts_ERCC"
## [59] "log10_total_counts_ERCC"
## [60] "pct_counts_ERCC"
## [61] "pct_counts_in_top_50_features_ERCC"
## [62] "pct_counts_in_top_100_features_ERCC"
## [63] "pct_counts_in_top_200_features_ERCC"
## [64] "pct_counts_in_top_500_features_ERCC"
# 可以发现细胞质控属性非常多,可以说是实用至极。
## 比如看每个样本测到的基因数量
tf <- sce$total_features_by_counts
boxplot(tf)
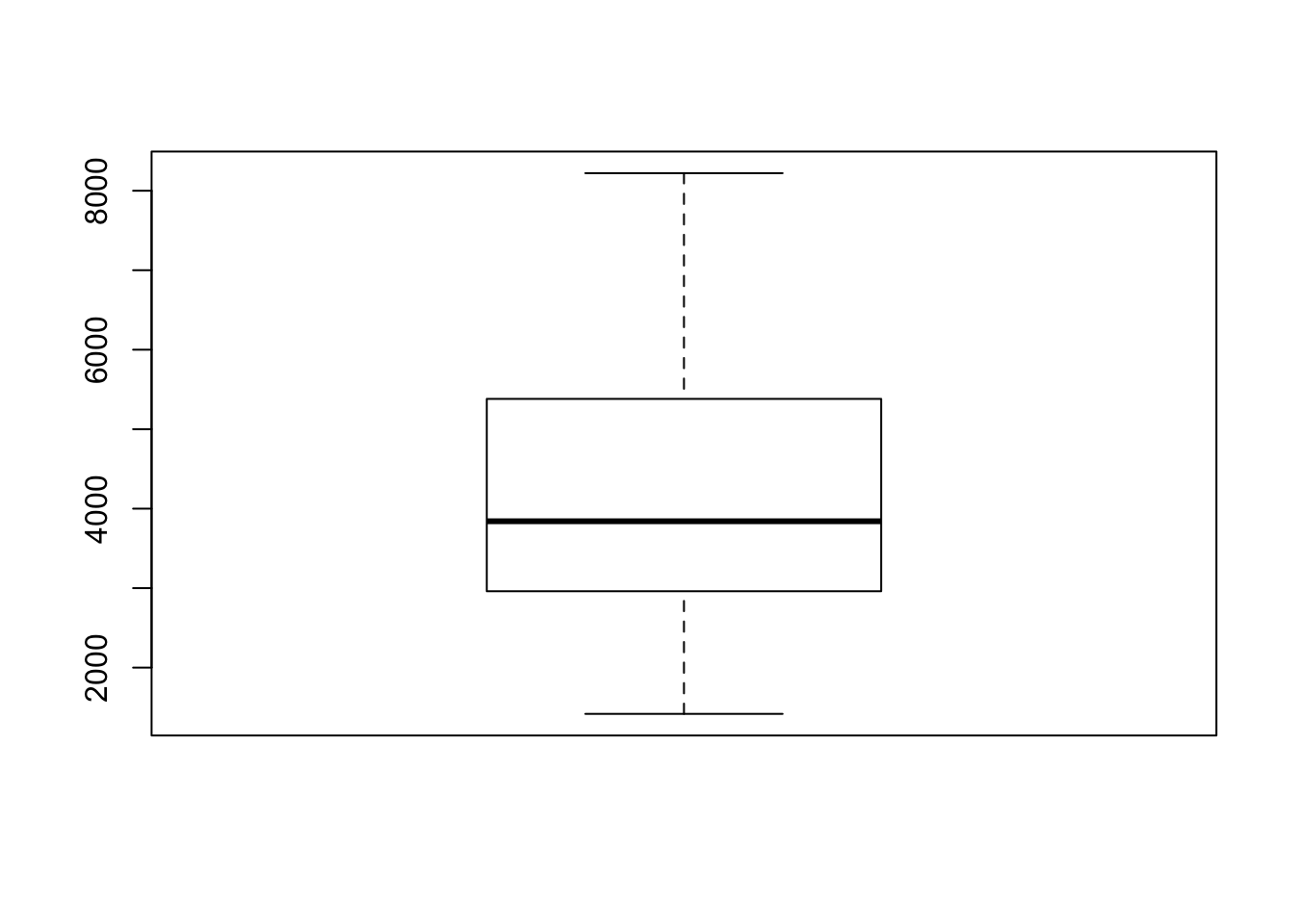
fivenum(tf)
## [1] 1418.0 2961.0 3841.5 5381.0 8221.0
还有很多其它指标,比如哪些细胞的ERCC含量过高,或者某些其高表达量基因占比太高,等等。
4.一些可视化
可视化函数非常多,所以这个R包才会被引用那么广泛,里面包括如下:
首先是一些细胞距离情况
- plotPCA: produce a principal components plot for the cells.
- plotTSNE: produce a t-distributed stochastic neighbour embedding (reduced dimension) plot for the cells.
- plotDiffusionMap: produce a diffusion map (reduced dimension) plot for the cells.
- plotMDS: produce a multi-dimensional scaling plot for the cells.
- plotReducedDim: plot a reduced-dimension representation of the cells.
然后是一些表达量相关的:
- plotExpression: plot expression levels for a defined set of features.
- plotPlatePosition: plot cells in their position on a plate, coloured by cell metadata and QC metrics or feature expression level.
- plotColData: plot cell metadata and QC metrics.
- plotRowData: plot feature metadata and QC metrics.
4.1 首先可视化表达量
可以添加一些细胞属性,展示如下:
# 挑选一些细胞属性(临床信息) 来进行可视化展示。
colnames(tmp)[25:28]
## [1] "Biological_Condition" "Coverage_Type" "Cluster1"
## [4] "Cluster2"
# 主要展现下面这些基因
rownames(sce)[1:6]
## [1] "A1BG" "A1BG-AS1" "A1CF" "A2M" "A2M-AS1" "A2ML1"
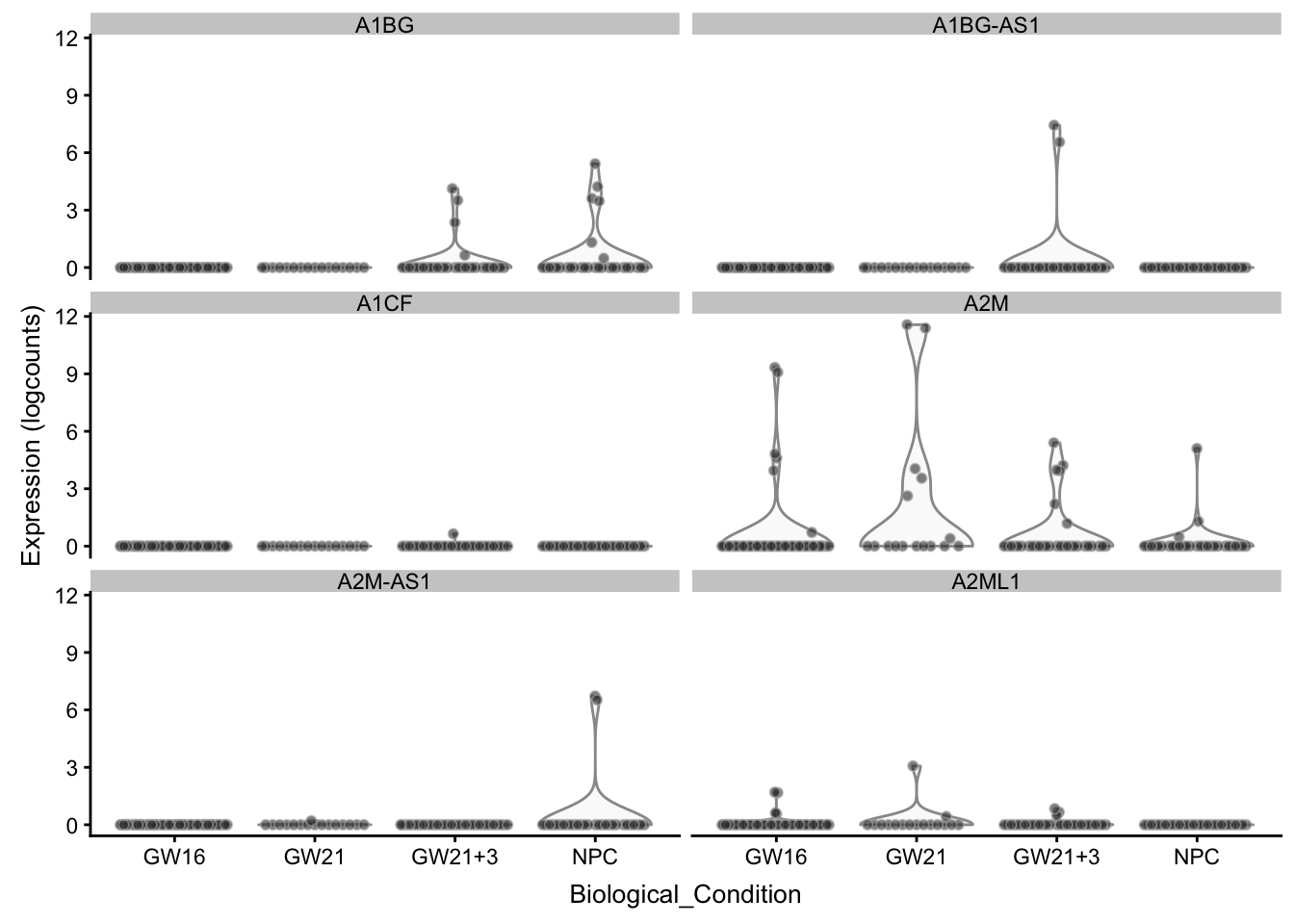
展示一些基因在不同细胞分类的表达区别
## --------------------
plotExpression(sce, rownames(sce)[1:6],
x = "Biological_Condition",
exprs_values = "logcounts")
散点图,展示两个基因的相关性,这里批量展示6对相关性
## -------------------------------------------
plotExpression(sce, rownames(sce)[1:6],
x = rownames(sce)[1])
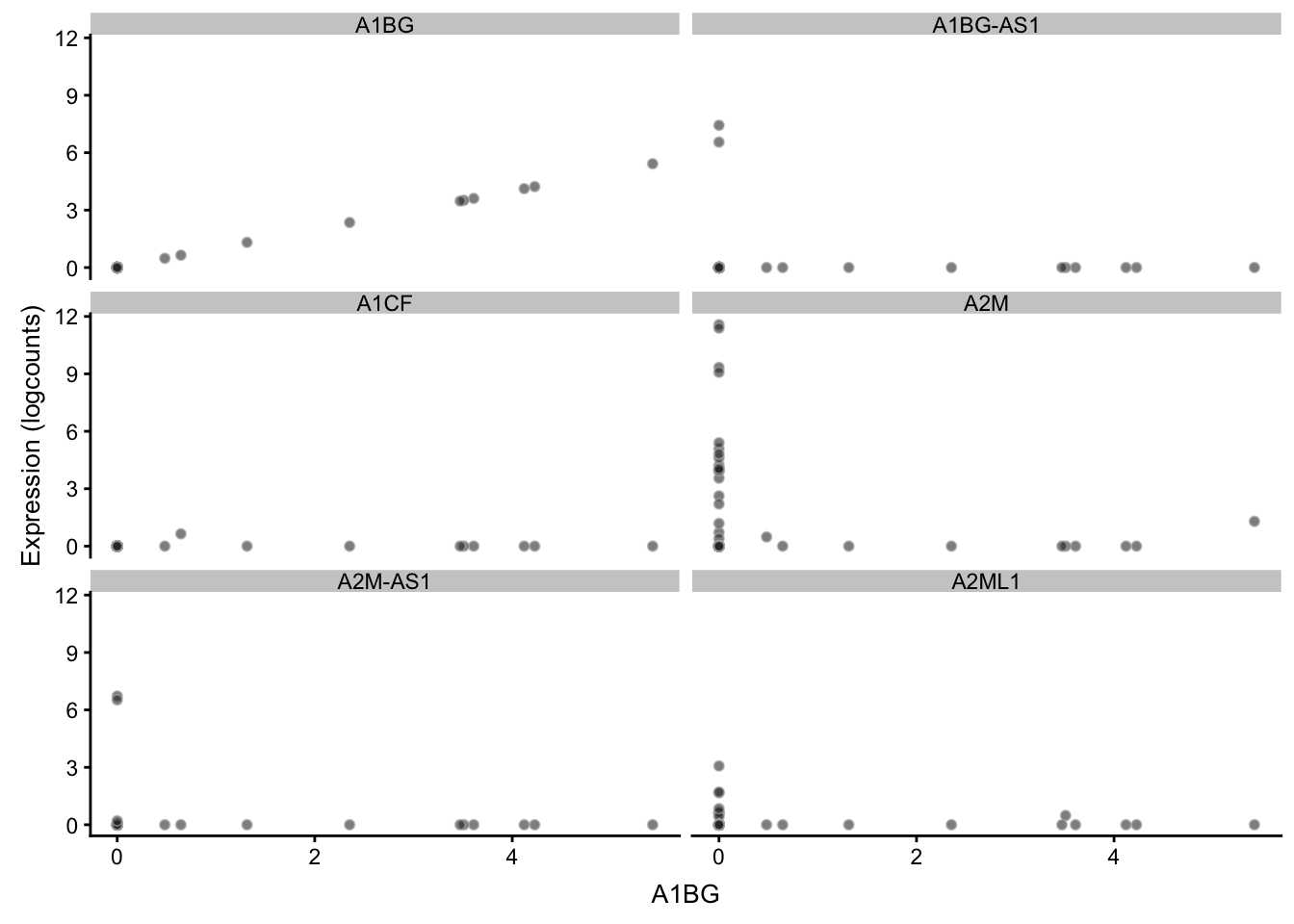
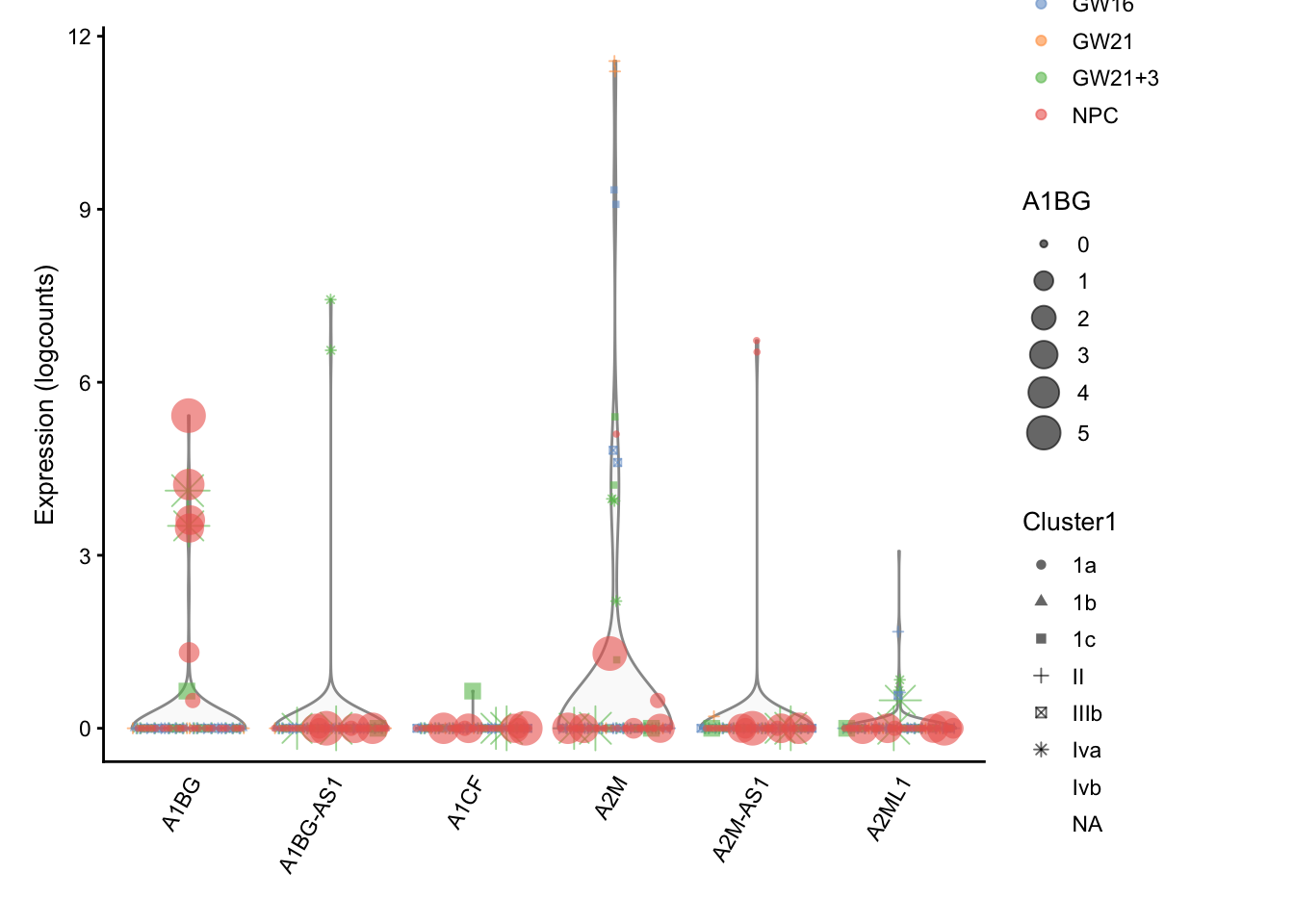
还可以更复杂
## ---------------
plotExpression(sce, rownames(sce)[1:6],
colour_by = "Biological_Condition",
shape_by = "Cluster1",
size_by = rownames(sce)[1])
4.2 还可以可视化细胞距离分布
这里可以学习PCA或者tSNE的使用
# 举例
sce <- runPCA(sce)
# 这里并没有进行任何基因的挑选,就直接进行了PCA,与 seurat包不一样。
reducedDimNames(sce)
## [1] "PCA"
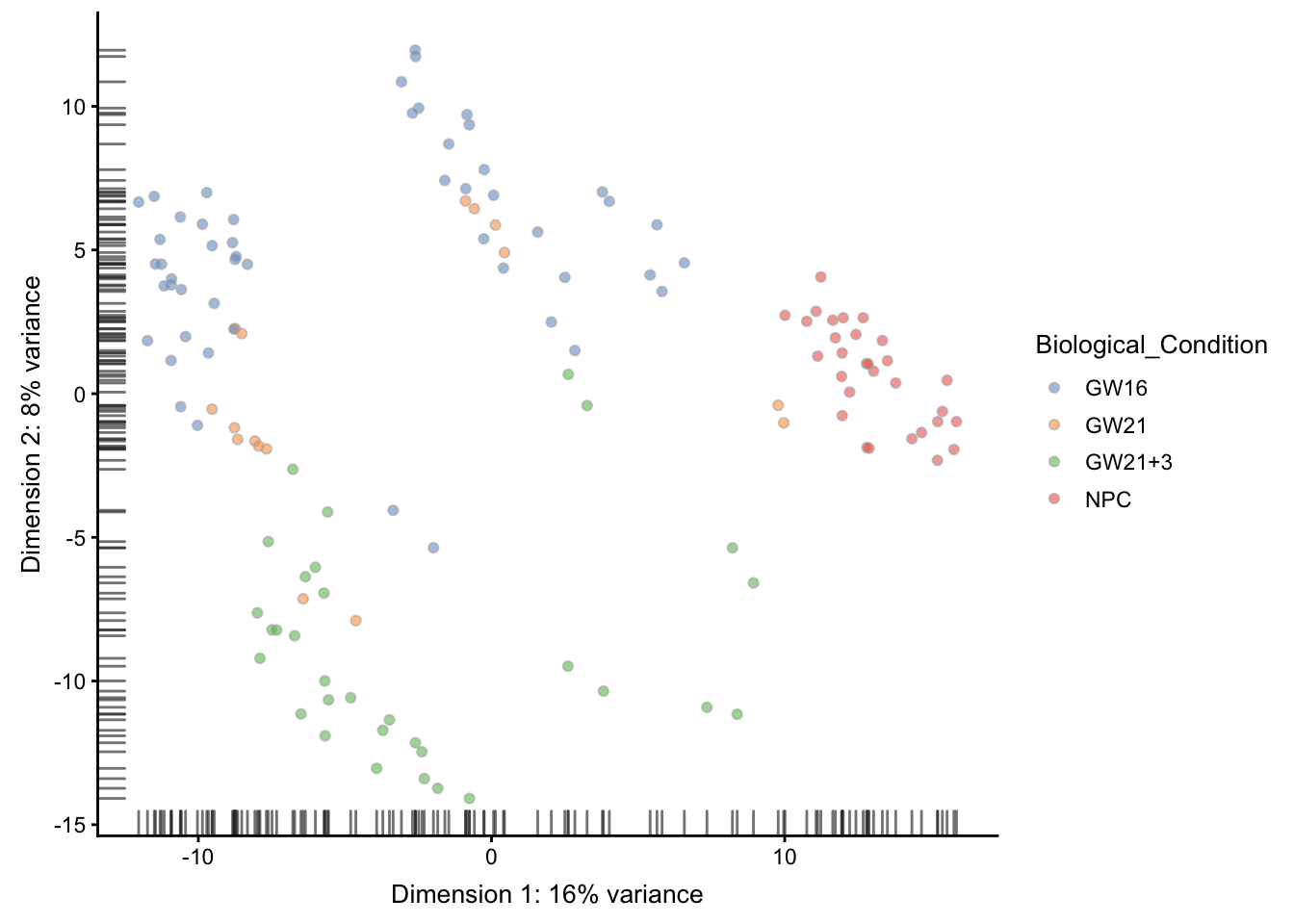
PCA分布图上面添加临床信息,同样的可以看到GW16混在了GW21里面
## --------------
plotReducedDim(sce, use_dimred = "PCA",
colour_by = "Biological_Condition" )
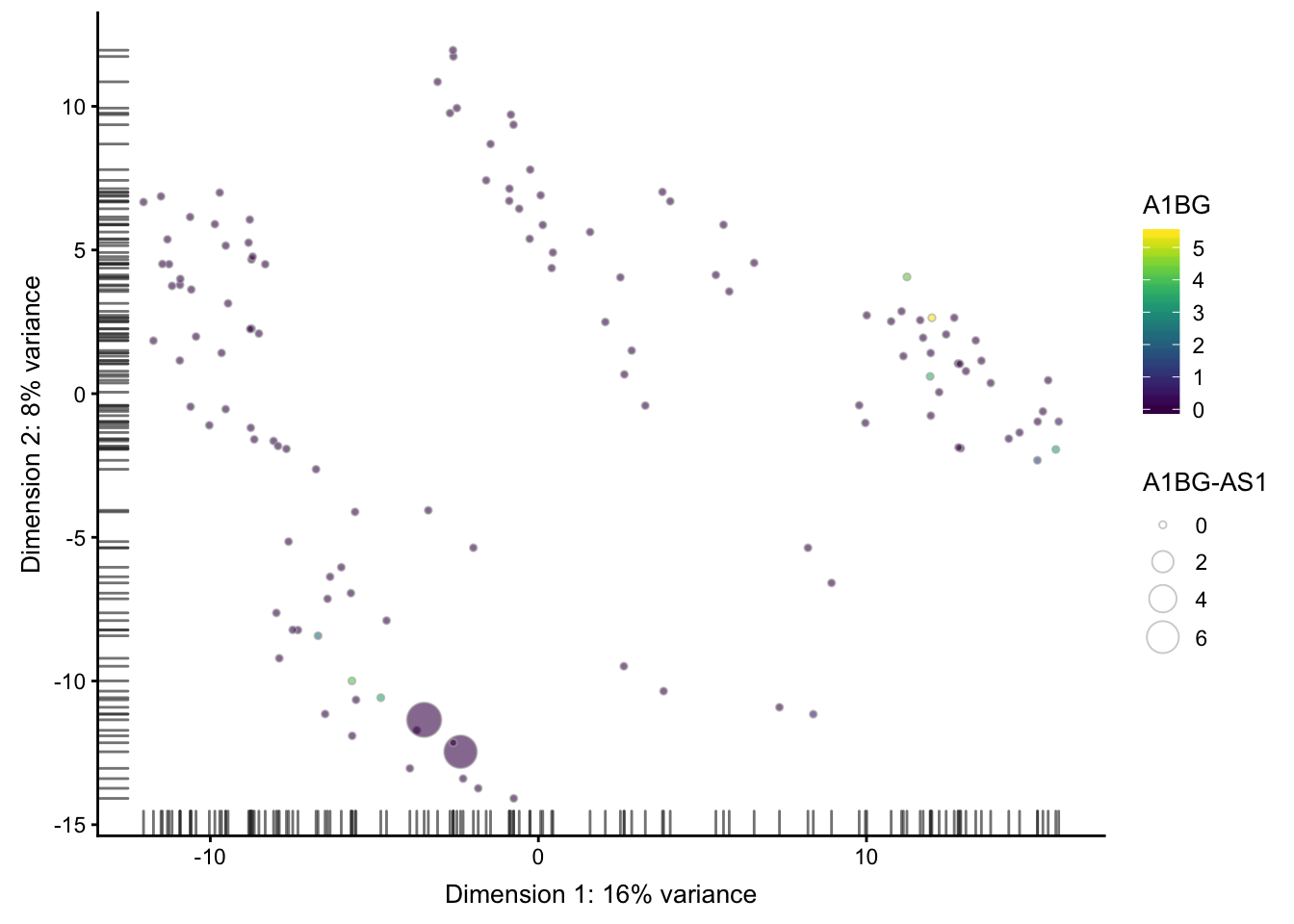
PCA分布图上面添加表达量信息, 但这个基本上不怎么用
## ------------
plotReducedDim(sce, use_dimred = "PCA",
colour_by = rownames(sce)[1],
size_by = rownames(sce)[2])
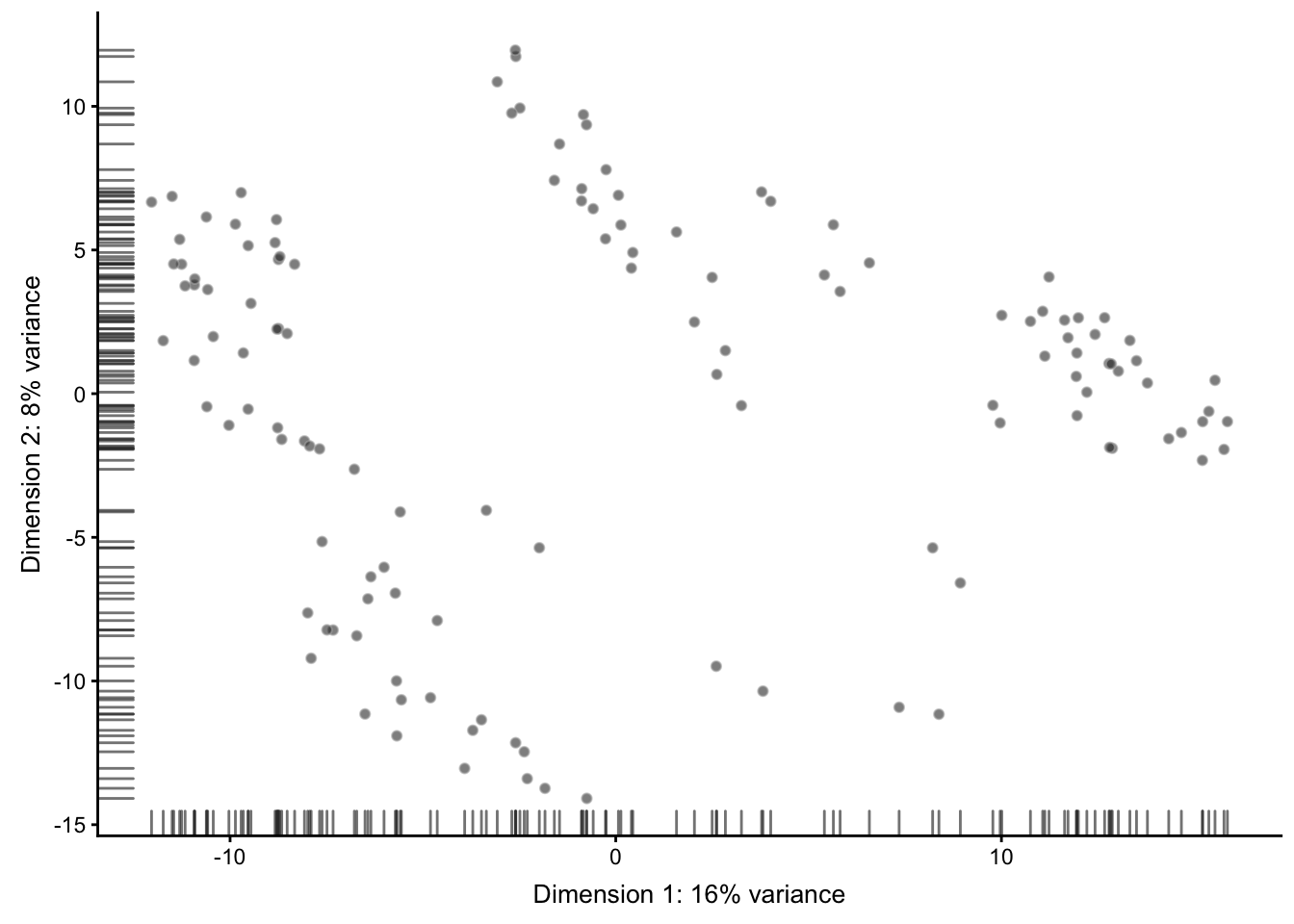
最原始的
## --------------
plotPCA(sce)
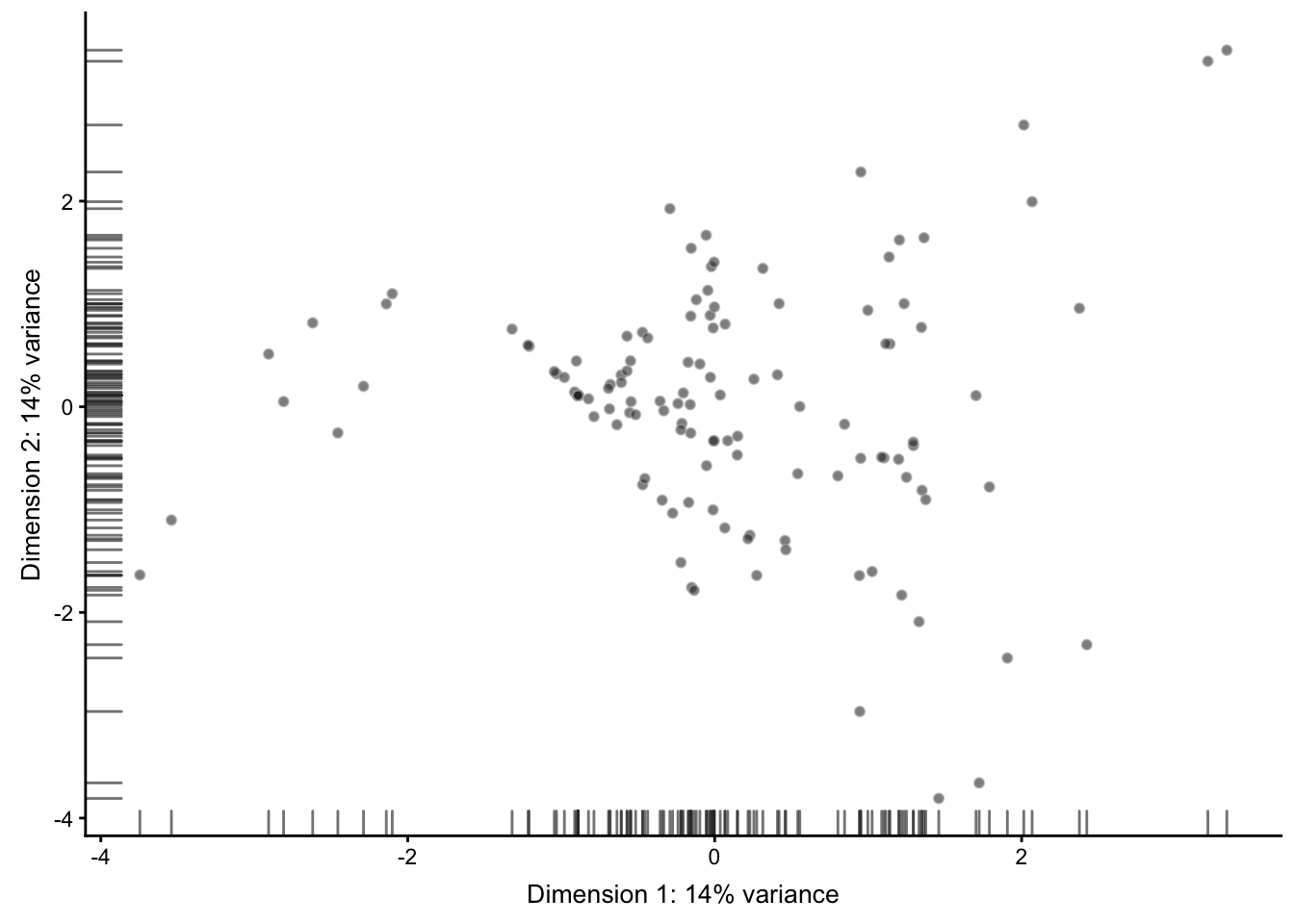
4.3 其它降维算法结果的可视化
## ----plot-pca-feature-controls---------------------------------------------
# 这里在真实场景中应用比较多。
sce2 <- runPCA(sce,
feature_set = rowData(sce)$is_feature_control)
plotPCA(sce2)
仅仅是选取前20个PC, 可以看到绘图时候细胞分布并没有太大区别
## ----plot-pca-4comp-colby-shapeby------------------------------------------
sce <- runPCA(sce, ncomponents=20)
plotPCA(sce, colour_by = "Biological_Condition" )
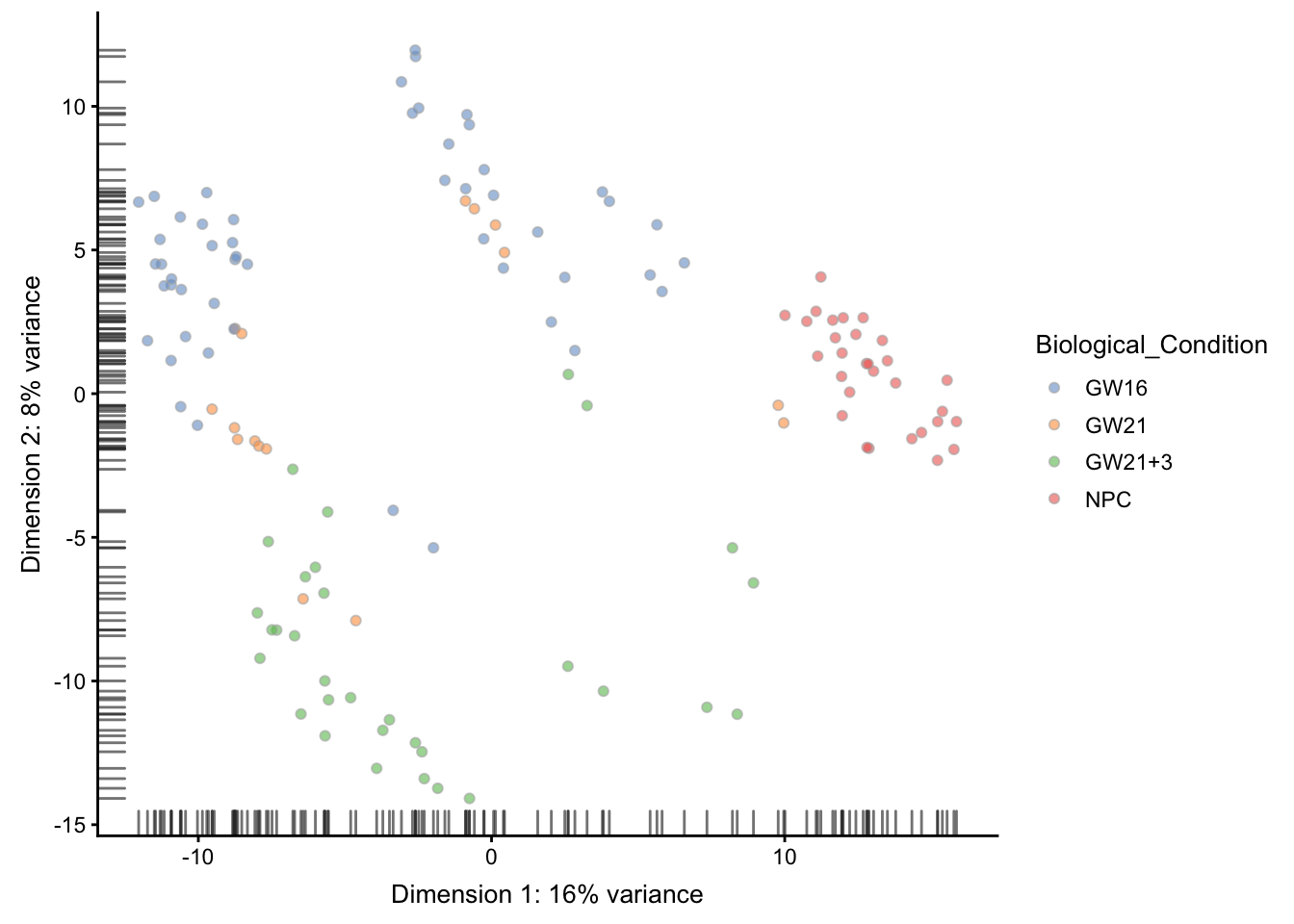
但是可以挑选指定的PC来可视化, 这里选择的是第4个PC
plotPCA(sce, ncomponents = 4,
colour_by = "Biological_Condition" )
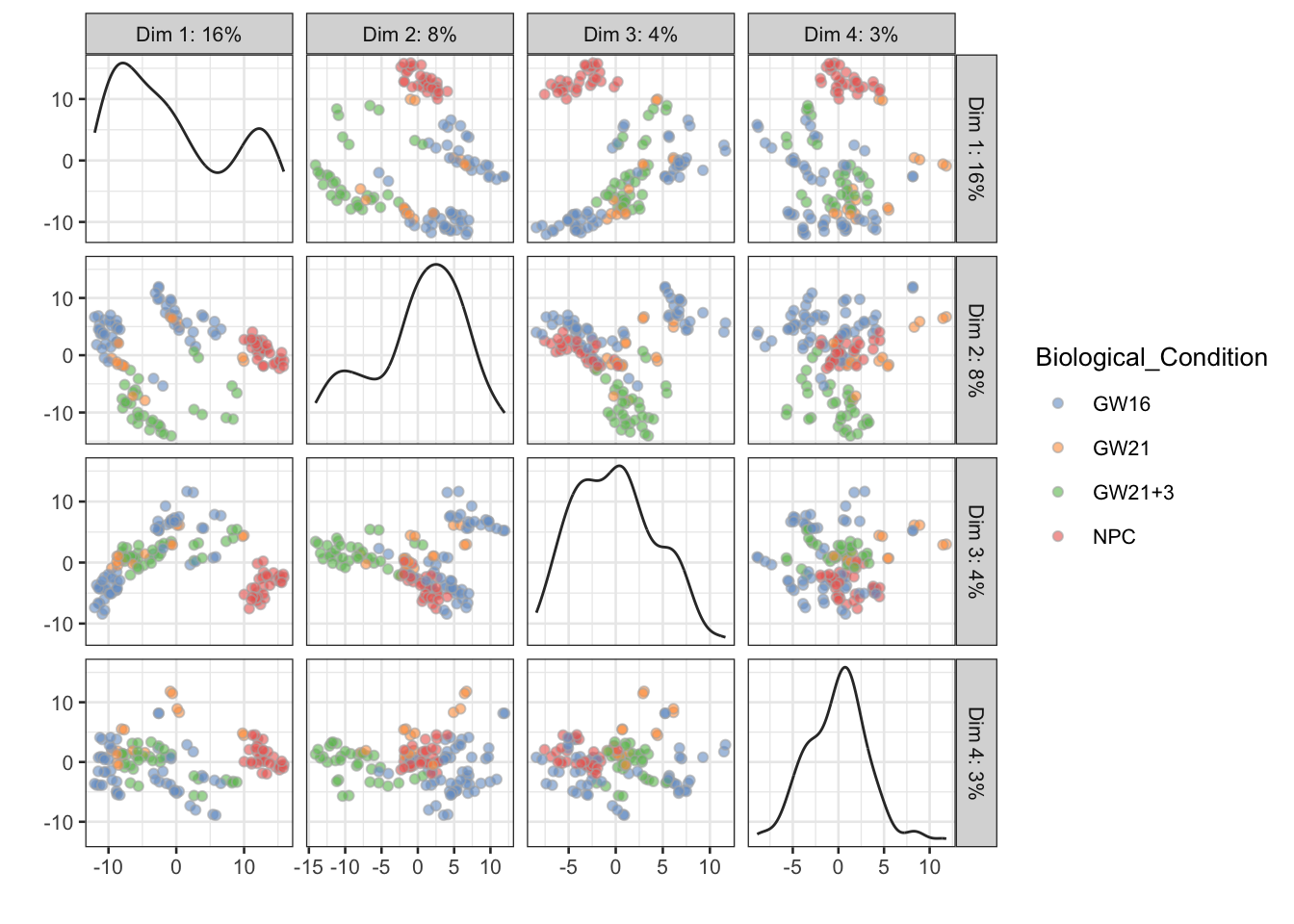
意义不大的展示
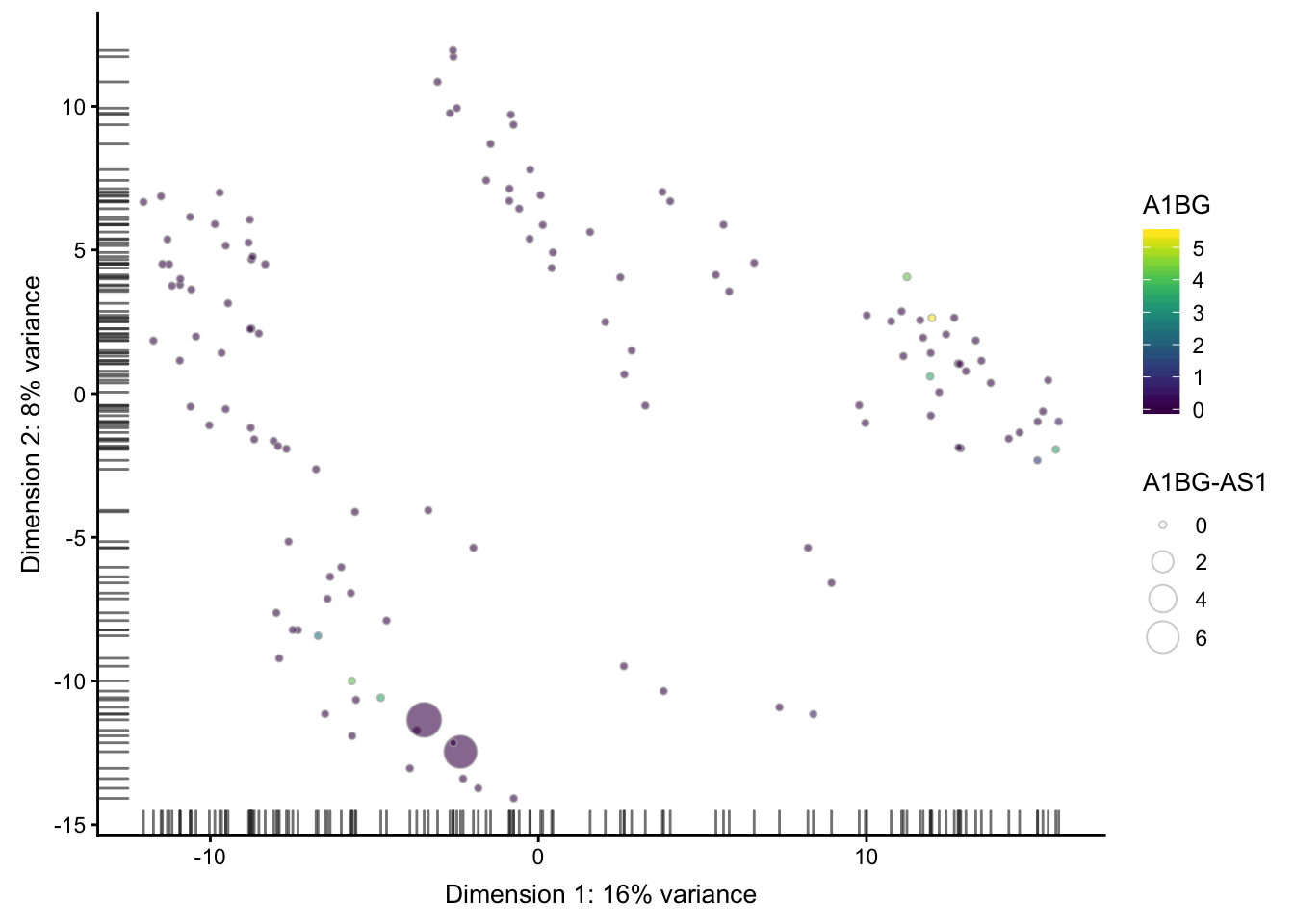
## ----plot-pca-4comp-colby-sizeby-exprs-------------------------------------
plotPCA(sce,
colour_by = rownames(sce)[1],
size_by = rownames(sce)[2])
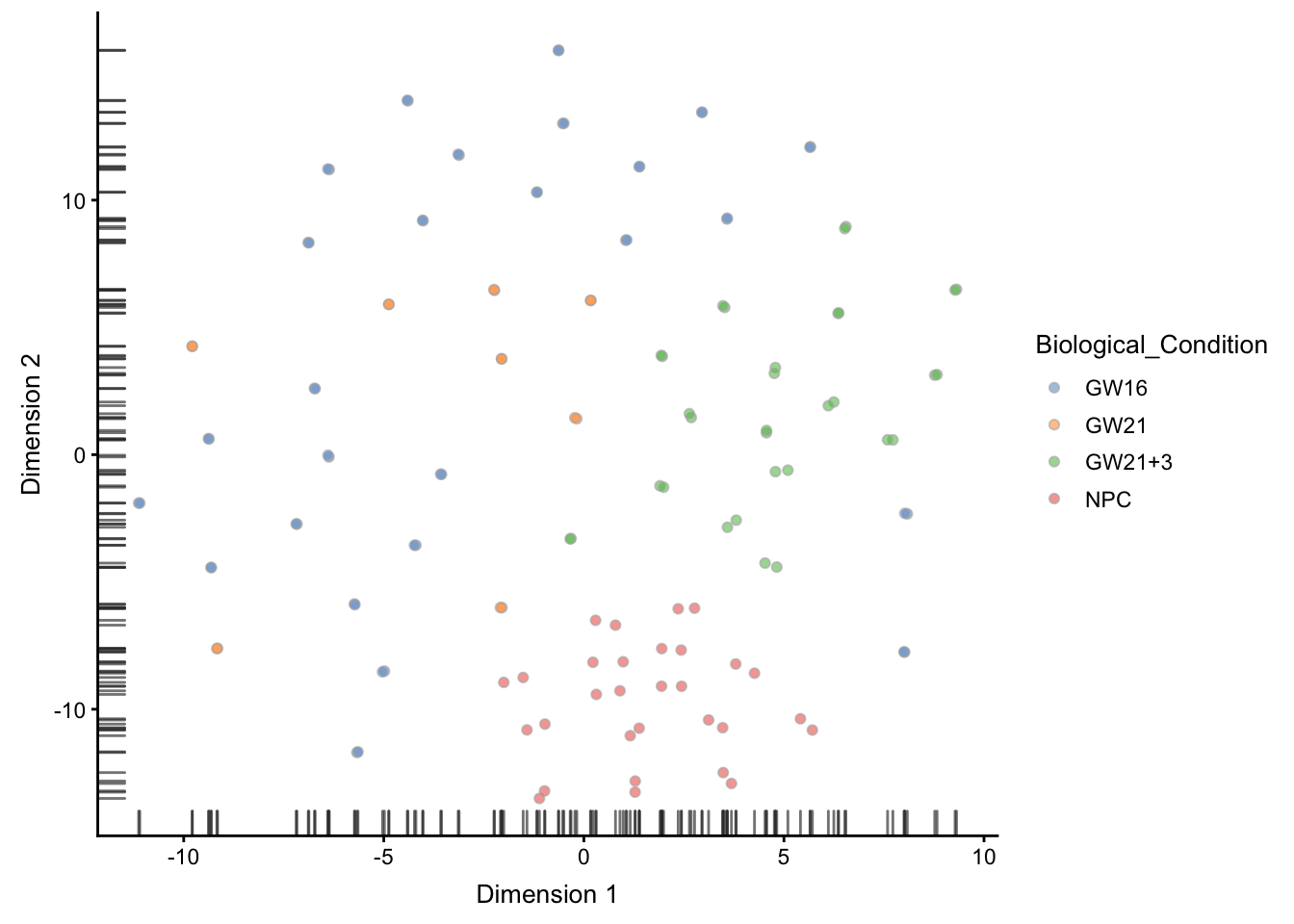
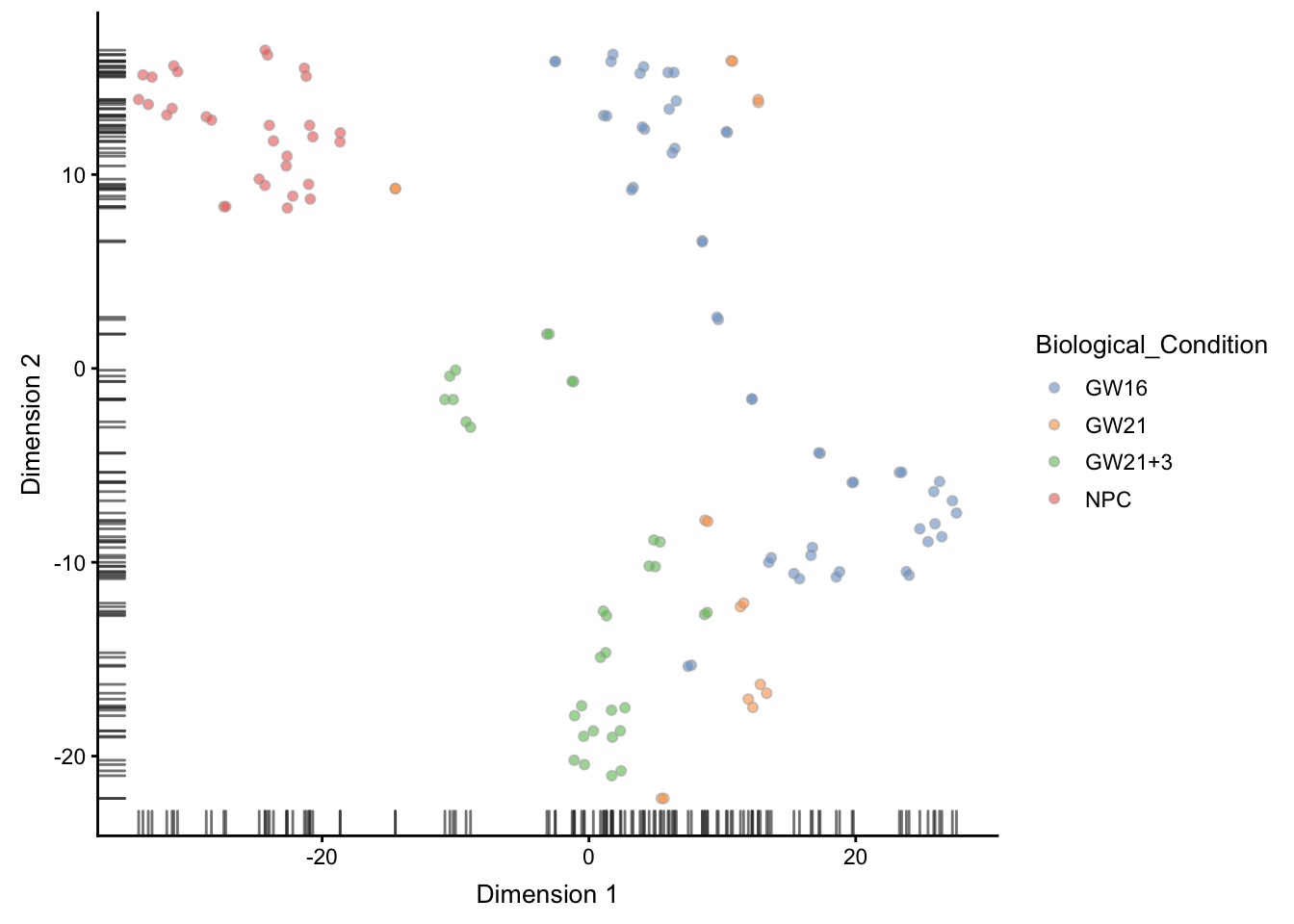
tSNE可视化
## ----plot-tsne-1comp-colby-sizeby-exprs------------------------------------
# Perplexity of 10 just chosen here arbitrarily.
set.seed(1000)
# 这里的这个 perplexity 参数很重要
sce <- runTSNE(sce, perplexity=30)
plotTSNE(sce,
colour_by = "Biological_Condition" )
## ----plot-tsne-from-pca----------------------------------------------------
set.seed(1000)
sce <- runTSNE(sce, perplexity=10, use_dimred="PCA", n_dimred=10)
plotTSNE(sce,
colour_by = "Biological_Condition" )
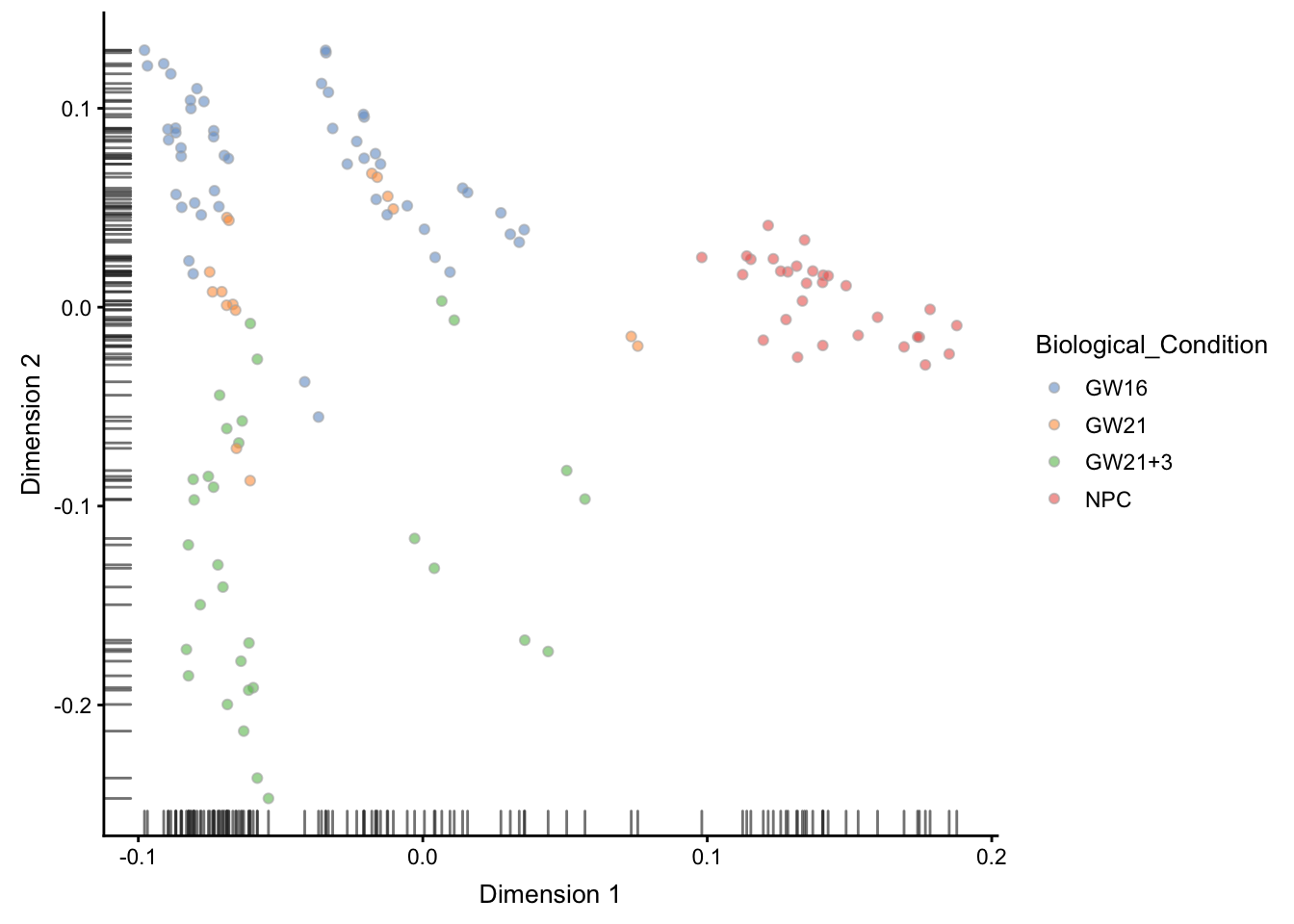
这一步会调用destiny
## ----plot-difmap-1comp-colby-sizeby-exprs----------------------------------
sce <- runDiffusionMap(sce)
plotDiffusionMap(sce,
colour_by = "Biological_Condition" )
5.关于SC3包
虽然 SC3包 跟 scater 联系很紧密,但毕竟不属于其知识点,这里就简单运行即可。
预估亚群, 这里显示有24个, 比较多
library(SC3) # BiocManager::install('SC3')
sce <- sc3_estimate_k(sce)
metadata(sce)$sc3$k_estimation
## [1] 24
rowData(sce)$feature_symbol <- rownames(rowData(sce))
一步运行sc3的所有分析, 相当耗费时间
这里kn表示的预估聚类数, 考虑到数据集是已知的,我们强行设置为4组, 具体数据要具体考虑。
# 耗费时间
kn <- 4 ## 这里可以选择 3:5 看多种分类结果。
sc3_cluster <- "sc3_4_clusters"
Sys.time()
## [1] "2019-03-11 17:44:18 CST"
sce <- sc3(sce, ks = kn, biology = TRUE)
Sys.time()
## [1] "2019-03-11 17:45:06 CST"
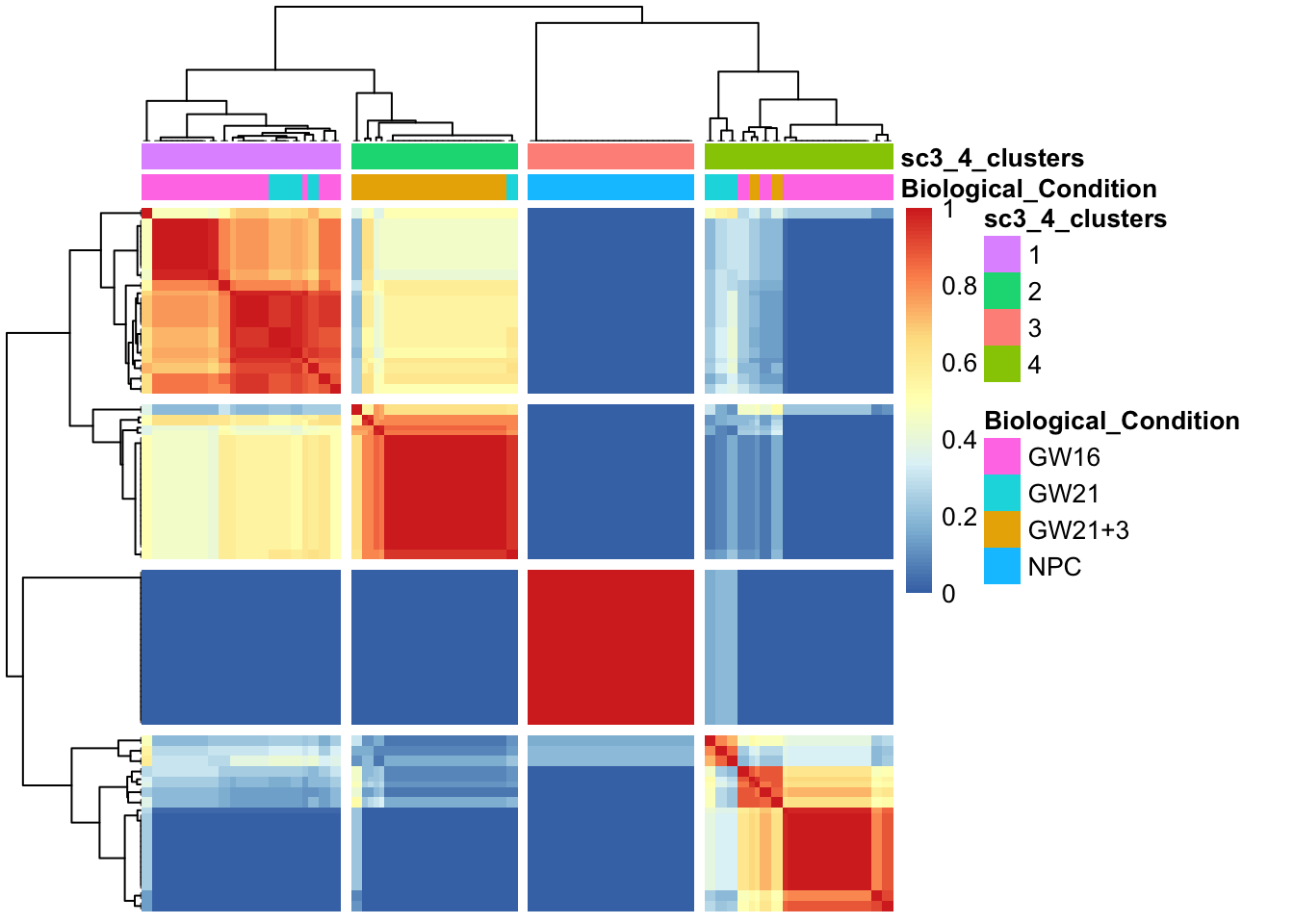
5.1 可视化展示部分, kn就是聚类数
热图: 比较先验分类和SC3的聚类的一致性
sc3_plot_consensus(sce, k = kn, show_pdata = c("Biological_Condition",sc3_cluster))
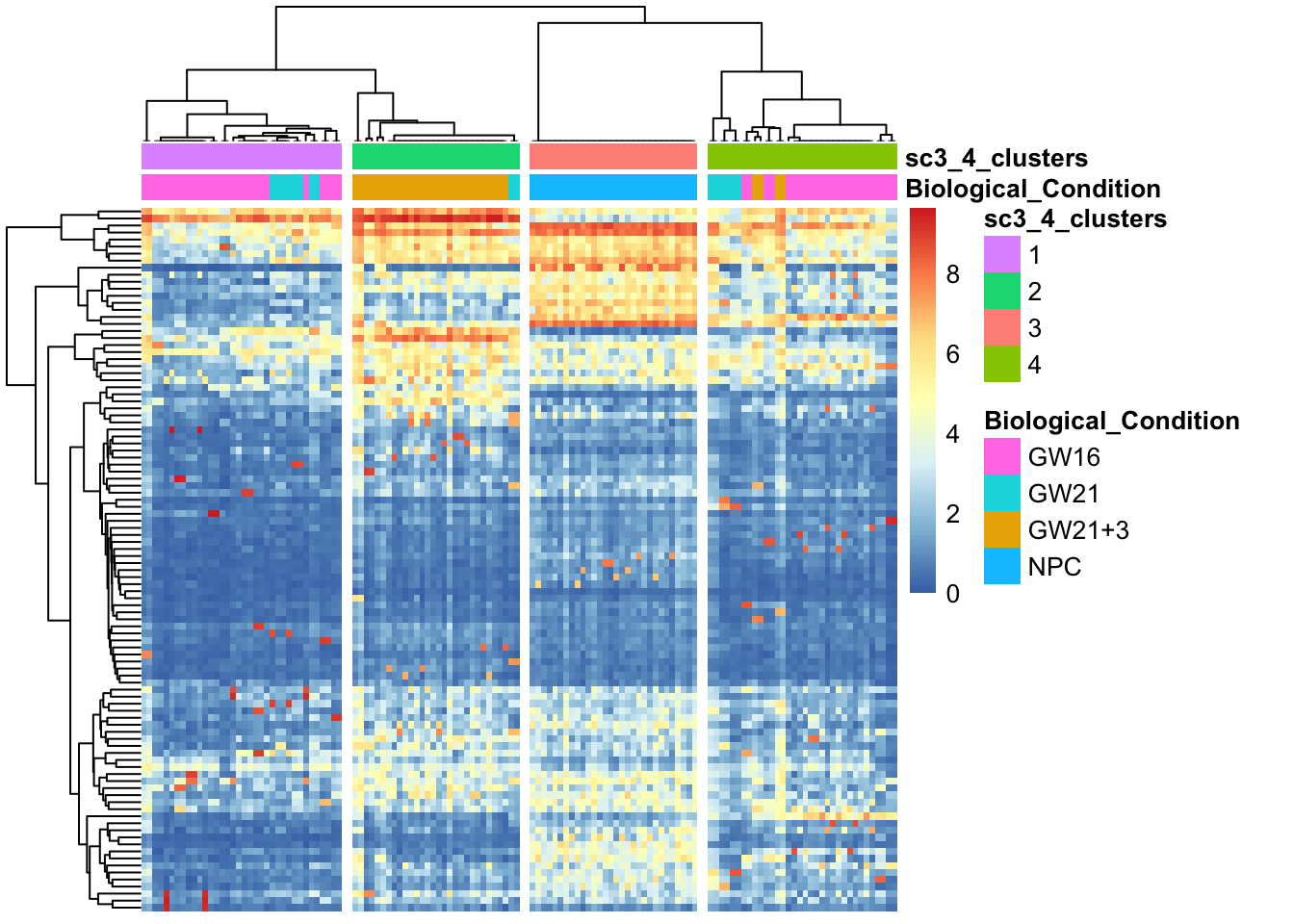
展示表达量信息
sc3_plot_expression(sce, k = kn, show_pdata = c("Biological_Condition",sc3_cluster))
展示可能的标记基因
sc3_plot_markers(sce, k = kn, show_pdata = c("Biological_Condition",sc3_cluster))
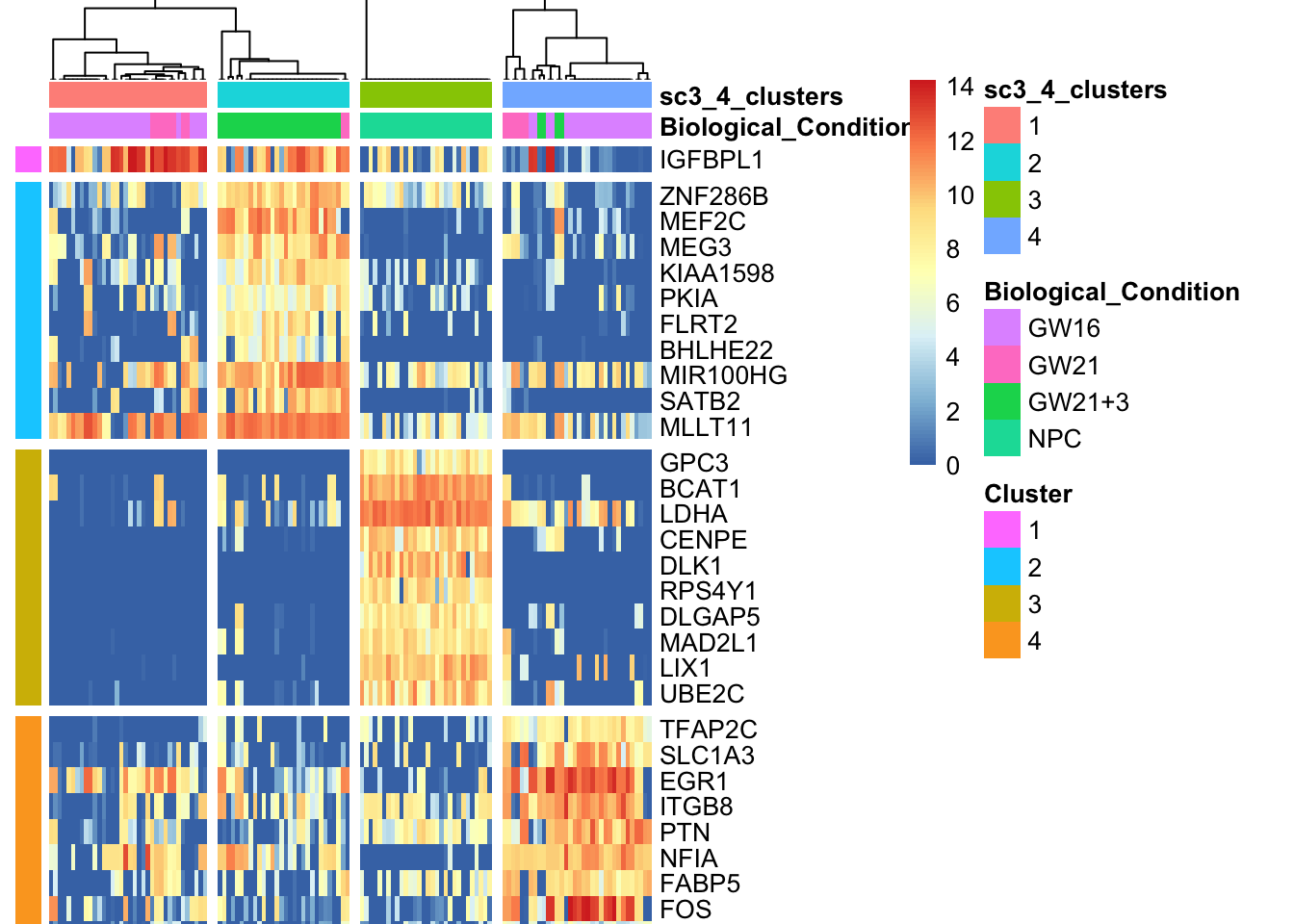
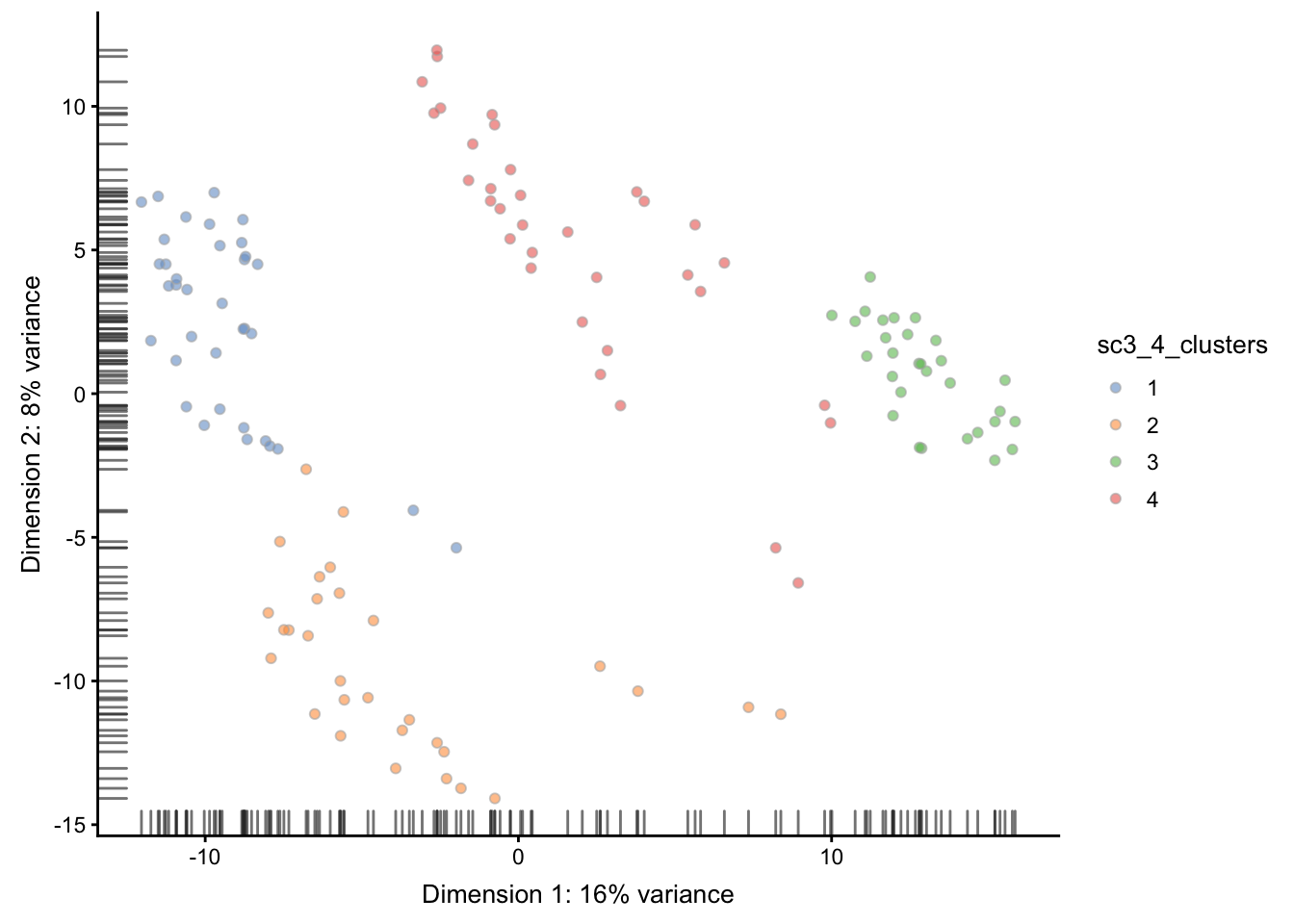
在PCA上展示SC3的聚类结果
plotPCA(sce, colour_by = sc3_cluster )
# sc3_interactive(sce)