1.准备工作
1.1 项目介绍

A basic overview of Seurat that includes an introduction to:
- (1) QC and pre-processing,QC和数据预处理
- (2) Dimension reduction,降维
- (3) Clustering,聚类
- (4) Differential expression,差异分析
1.2 数据来源及下载
本文的范例数据为seurat官网的pbmc-3k数据,可以从官网直接下载:raw data
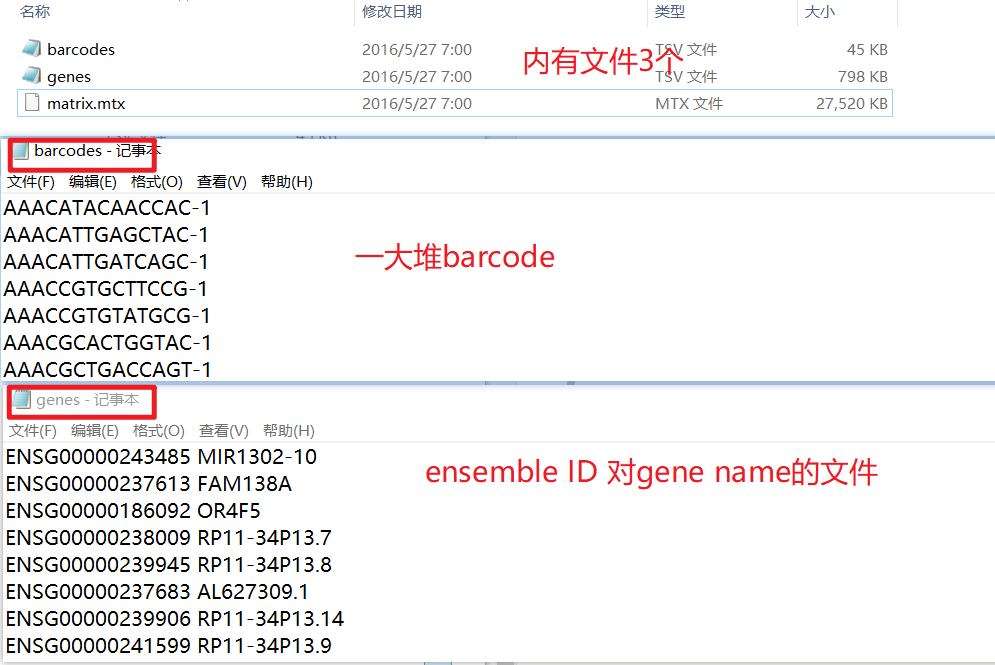
- 文件1:barcodes
- 文件2:ensemble ID to gene name
- 文件3:真正的测序数据
1.3 R包安装
# 安装Seurat包
install.packages('BiocManager') # 没有的话就安装一个
BiocManager::install('multtest')
install.packages('Seurat')
install.packages("patchwork")
library(dplyr)
library(Seurat)
library(patchwork)
# 加载Seurat包
library(Seurat)
1.4 读入数据
# 读入pbmc数据
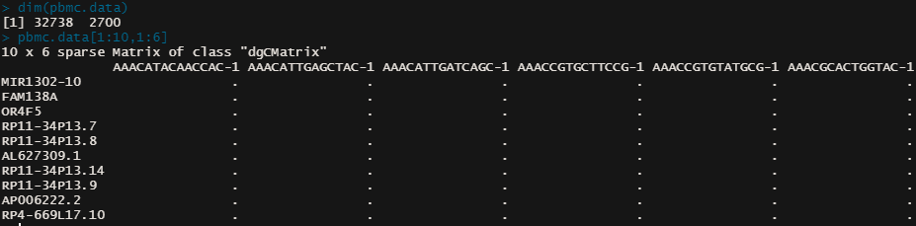
pbmc.data <- Read10X(data.dir = "./Seurat_practise/hg19/")
# 查看稀疏矩阵的维度,即基因数和细胞数
dim(pbmc.data)
# 预览稀疏矩阵(1~10行,1~6列)
pbmc.data[1:10,1:6]
2. 创建Seurat对象与数据过滤
2.1 创建Seurat对象并并设置条件筛选细胞
- (1)能检测到某个基因的细胞数,即unique基因的分布情况,对应上面的min.cells = 3;
- (2)每个细胞能检测到的基因数,对应上面的min.features = 200;
- (3)线粒体gene的比例要足够小,使用PercentageFeatureSet函数计算,以MT-开头的则是线粒体gene。
pbmc <- CreateSeuratObject(counts=pbmc.data,
project="pbmc2700",
min.cells = 3,min.features = 200)
# 举例查看数据
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
在使用CreateSeuratObject()创建对象的同时,过滤数据质量差的细胞。保留在>=3个细胞中表达的基因;保留能够检测到>=200个基因的细胞。
2.2 添加线粒体百分比列
# 计算每个细胞的线粒体基因转录本数的百分比(%),使用[[]]操作符放到metadata中
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc,pattern = "^MT-")
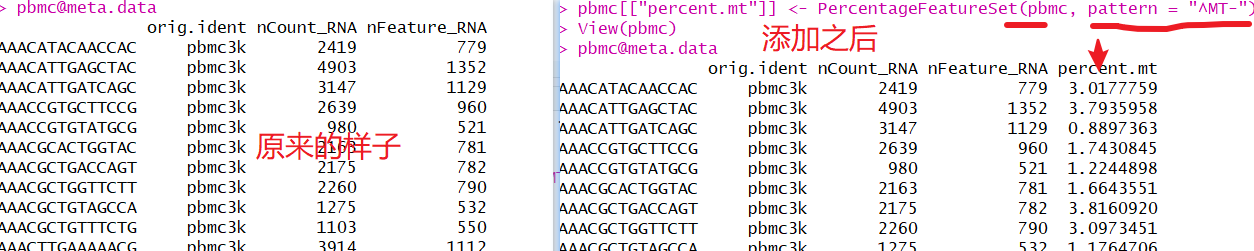
2.3 可视化QC矩阵
把nFeature,nCount,还有MT percentage画在同一个图上,检查数据情况。
# 使用小提琴图可视化QC 矩阵
VlnPlot(pbmc,
features = c("nFeature_RNA",
"nCount_RNA",
"percent.mt"),
ncol = 3)
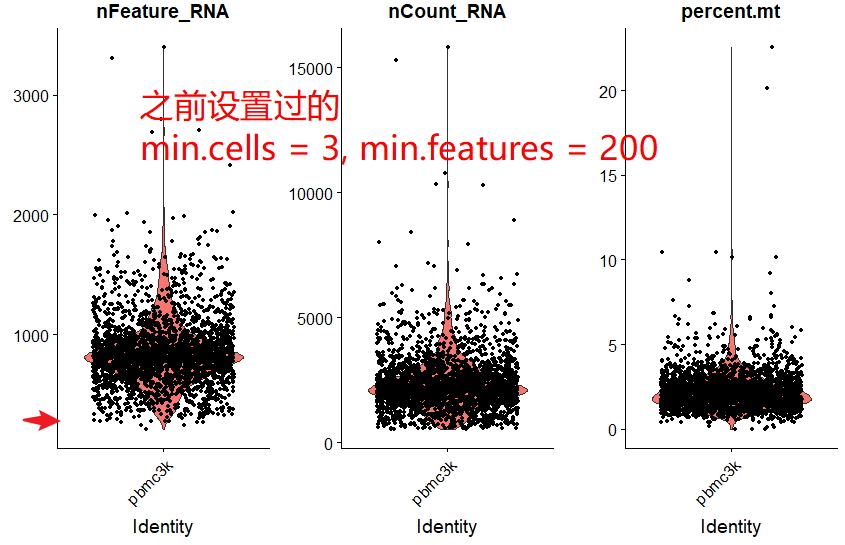
可以看到,细胞的unique gene(nFeature)和total molecular(nCount)都挺集中的,分别在1000和2000左右,而线粒体基因的比例在百分之2.5左右,合格标准大概设置为多少,需要查一下。
2.4 查看不同特征之间的关系
FeatureScatter顾名思义,就是做Feature的Scatter散点图,这个命令可以用于任何你感兴趣的feature之间的interaction探索。
plot1 <- FeatureScatter(pbmc,feature1="nCount_RNA",
feature2="percent.mt")+NoLegend()
plot2 <- FeatureScatter(pbmc,feature1="nCount_RNA",
feature2="nFeature_RNA")+NoLegend()
CombinePlots(plots=list(plot1,plot2))
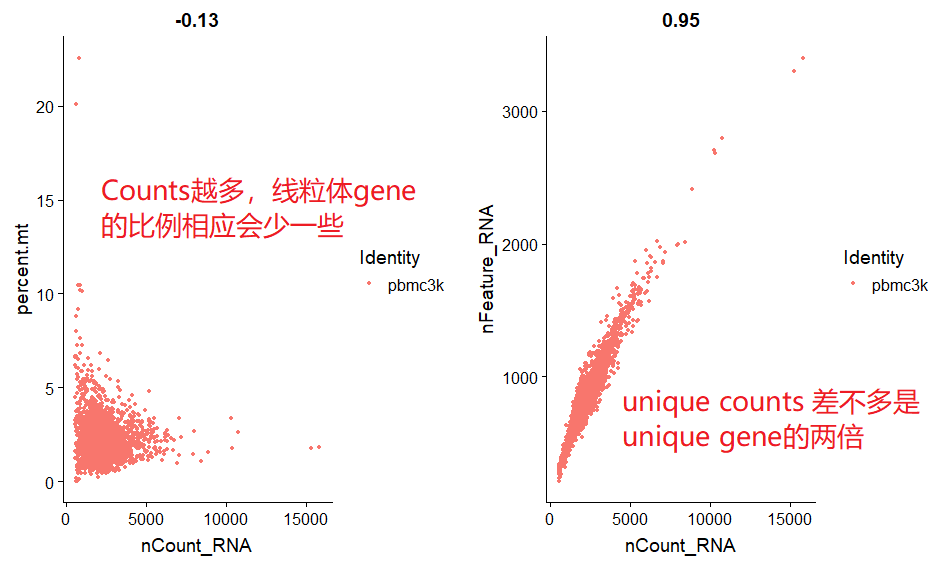
Note:和”nCount_RNA”为细胞的总RNA分子数;nFeature_RNA为细胞的unique基因数。
2.5 设定标准,筛选数据
过滤细胞:保留gene数大于200小于2500的细胞;目的是去掉空GEMs和1个GEMs包含2个以上细胞的数据;而保留线粒体基因的转录本数低于5%的细胞,为了过滤死细胞等低质量的细胞数据
pbmc <- subset(pbmc,subset=nFeature_RNA>200 &
nFeature_RNA<2500 &
percent.mt<5)
3.表达量数据标准化(Normalizing the data)
从数据集中删除不需要的单元格后,下一步是标准化数据。默认情况下,使用全局缩放规范化方法LogNormalize,该方法通过总表达式对每个单元格的特征表达式度量进行标准化,并将其乘以一个缩放因子(默认为10,000),然后对结果进行log转换。标准化值存储在pbmc[[“RNA”]]@data中。
#表达量数据标准化:LogNormalize的算法:A=log(1+(UMI_A/UMI_Total)*10000)
pbmc <- NormalizeData(pbmc,normalization.method = "LogNormalize",
scale.factor = 10000)
# 在默认设定的情况下,上面的的代码可简写为:
pbmc <- NormalizeData(pbmc)
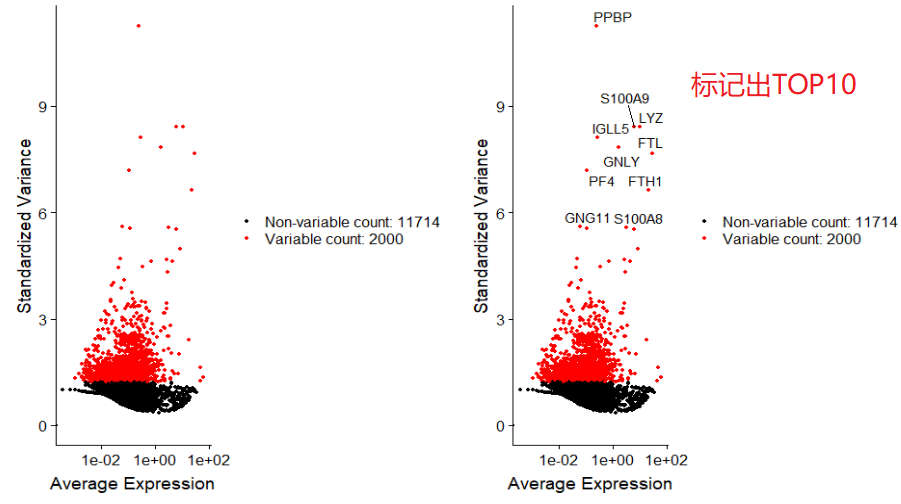
4.鉴定表达高变基因(Feature selection)
使用FindVariableFeatures完成差异分析,选择数据集中差异较高的特征基因(默认2000)并用于下游分析。
# 鉴定表达高变基因(2000个),用于下游分析,如PCA;
pbmc <- FindVariableFeatures(pbmc,selection.method = "vst",
nfeatures = 2000)
# 提取表达量变化最高的10个基因;
top10 <- head(VariableFeatures(pbmc),10)
top10

plot3 <- VariableFeaturePlot(pbmc)+NoLegend()
plot4 <- LabelPoints(plot=plot3,
points=top10,
repel=TRUE,
xnudge = 0,ynudge = 0)
plot4
5.缩放数据(Scaling the data)
PCA分析数据准备,使用ScaleData()进行数据归一化;默认只是标准化高变基因(2000个),速度更快,不影响PCA和分群,但影响热图的绘制。
5.1 缩放的标准
ScaleData函数:
- (1)使每个基因在所有细胞间的表达量均值为0
- (2)使每个基因在所有细胞间的表达量方差为1
缩放操作给予下游分析同等的权重,这样高表达基因就不会占主导地位,其结果存储在pbmc[[“RNA”]]@scale.data中**
5.2 对所有基因进行缩放
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
缩放是Seurat工作流程中的一个重要步骤,但仅限于将要进行PCA的基因。因此,ScaleData中的默认值只是对前面确定的变量特性(默认值为2,000)执行缩放。要做到这一点,可以忽略前面函数调用中的features参数,即pbmc <- ScaleData(pbmc)。
由于Seurat heatmap(与DoHeatmap一起产生,如下图所示),需要对heatmap中所有基因进行缩放,以确保高表达的基因不会在heatmap中占主导地位。为了确保我们在后面的热图中不会遗漏任何基因,我们将缩放所有基因。
5.3 删除不需要的变量
pbmc <- ScaleData(pbmc,vars.to.regress = "percent.mt")
使用ScaleData函数从单细胞数据集中删除不需要的变量。例如,我们可以“regress out”与细胞周期阶段或线粒体污染相关的异质性(去除不需要的变量,即校正协变量,校正线粒体基因比例的影响)。
6.线性降维(PCA)
# 线性降维(PCA),默认用高变基因集,但也可通过features参数自己指定;
pbmc <- RunPCA(pbmc,features=VariableFeatures(object=pbmc))
# 检查PCA分群结果,这里只展示前12个PC,每个PC只显示3个基因;
print(pbmc[["pca"]],dims=1:12,nfeatures = 3)
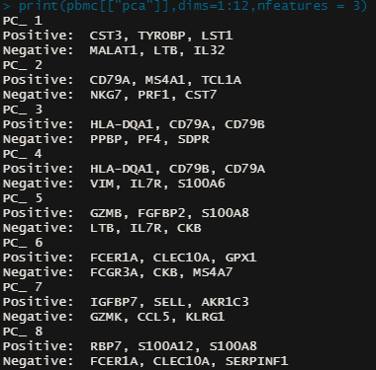
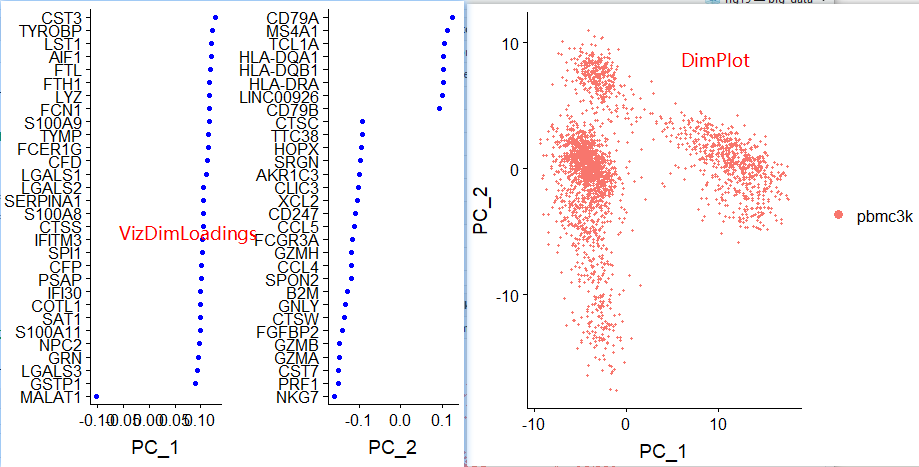
Seurat支持多种方式来可视化PCA 数据:VizDimReduction,DimPlot,DimHeatmap。
# VizDimReduction,小范围试一下
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
# 绘制PCA散点图;
DimPlot(pbmc,reduction = "pca")+NoLegend()
DimHeatmap可以探索数据集的异质性来源,帮助确定用于下游分析的主成分。细胞和features都是根据它们的PCA分数排序。
DimHeatmap(pbmc, dims = 1,
cells = 500, balanced = TRUE)
DimHeatmap(pbmc, dims = 1:15,
cells = 500, balanced = TRUE)

可以看到PC1-6对于数据分离比较有帮助,而越往后面帮助越小,可以通过这种方式来确定,下游分析中用于分析的主成分。
7.确定数据集的分群个数
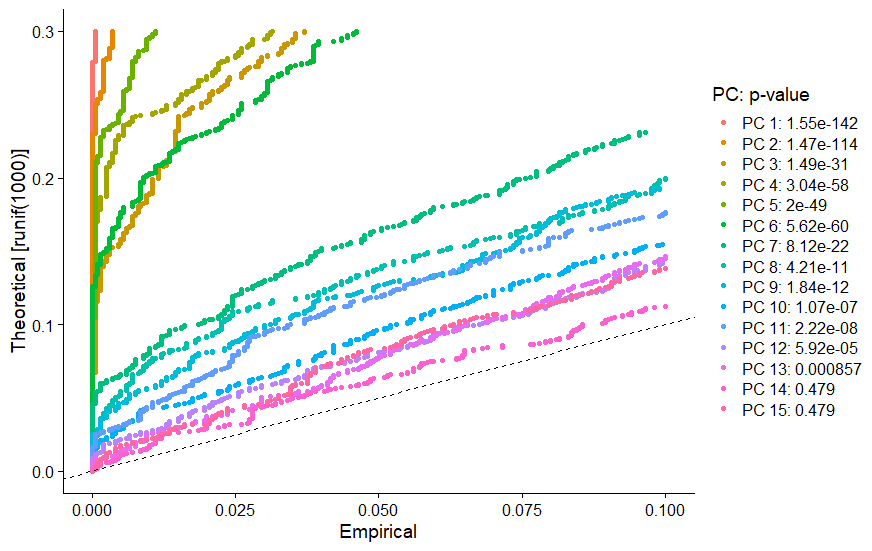
# 方法1:Jackstraw置换检验算法:重复取样(原数据的1%),重跑PCA,鉴定p-value较小的PC;计算’null distribution‘(即零假设成立时)时的基因scores;
pbmc <- JackStraw(pbmc,num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc,dims=1:20)
JackStrawPlot(pbmc,dims=1:15)
# 方法2:肘部图(碎石图),基于每个主成分对方差解释率的排名;
ElbowPlot(pbmc)
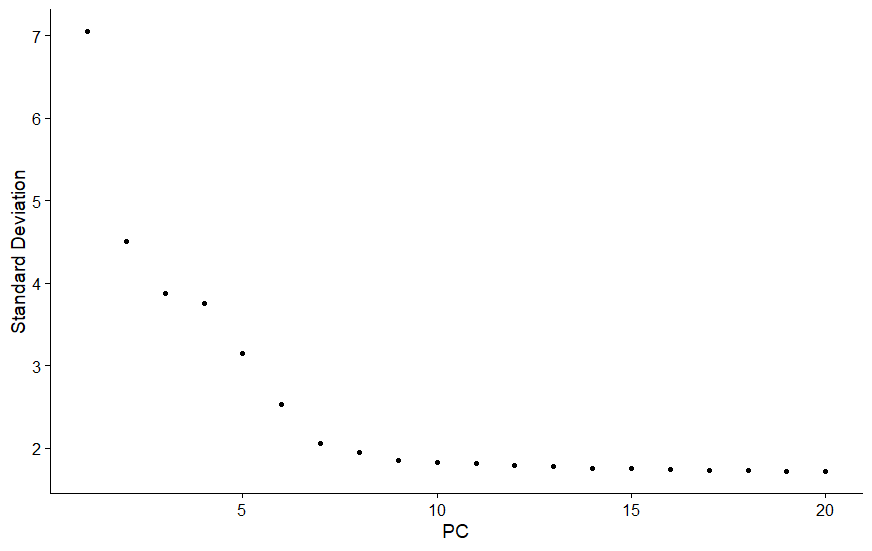
分群个数这里选择10,建议尝试选择多个主成分个数做下游分析,对整体影响不大;在选择此参数时,建议选择偏高的数字(“宁缺毋滥”,为了获取更多的稀有分群);有些亚群很罕见,如果没有先验知识,很难将这种大小的数据集与背景噪声区分开来。
8.细胞聚类(Cluster the cells)
# 基于PCA空间中的欧式距离计算nearest neighbor graph,优化任意两个细胞间的距离权重(输入上一步得到的PC维数);
pbmc <- FindNeighbors(pbmc, dims = 1:10) # 前10个PC
#接着优化模型,resolution参数决定下游聚类分析得到的分群数,对于3k左右的细胞,设为0.4-1.2能得到较好的结果(官方说明);如果数据量增大,该参数也应该适当增大;
pbmc <- FindClusters(pbmc, resolution = 0.5) # 分辨率是0.5
# 结果
# Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
# Number of nodes: 2638
# Number of edges: 95930
# 使用Ident()函数可查看不同细胞的分群;
head(Idents(pbmc), 8)

9.非线性降维(UMAP/tSNE)
Seurat提供了几种非线性降维的方法进行数据可视化(在低维空间把相似的细胞聚在一起),比如UMAP和t-SNE,运行UMAP需要先安装‘map-learn’包。
# 安装UMAP
reticulate::py_install(packages ="umap-learn")
# 用umap的方法,并可视化
pbmc <- RunUMAP(pbmc, dims = 1:10)
umapplot <- DimPlot(pbmc, reduction = "umap", pt.size=1)
umapplot
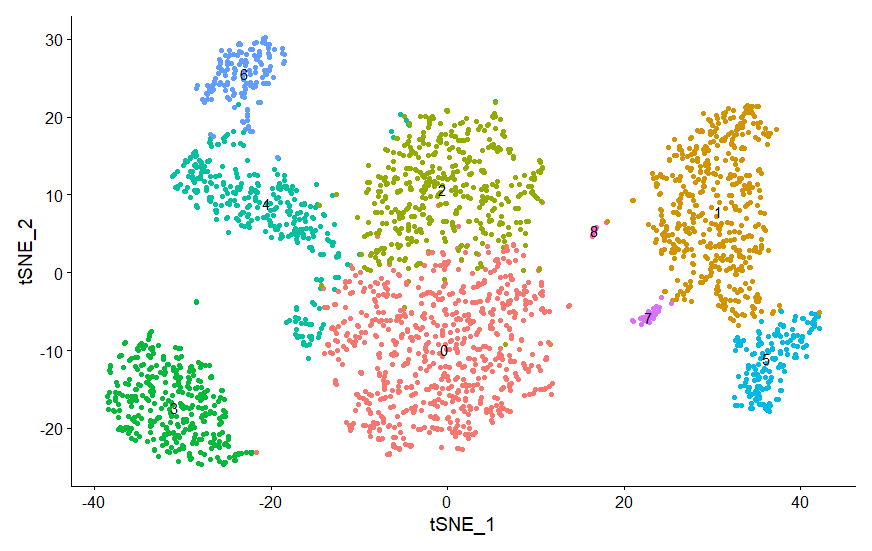
用DimPlot()函数绘制散点图,reduction=”tsne”,指定绘制类型;如果不指定,默认先从搜索umap,然后tsne,再然后pca;也可以直接使用这3个函数PCAPlot()、TSNEPlot()、UMAPPlot();cols、pt.size分别调整分组颜色和点的大小;
# 用tsne的方法,并可视化
pbmc <- RunTSNE(pbmc,dims=1:10)
tsneplot <- TSNEPlot(pbmc,label=TRUE,
pt.size=1.5)+NoLegend()
tsneplot
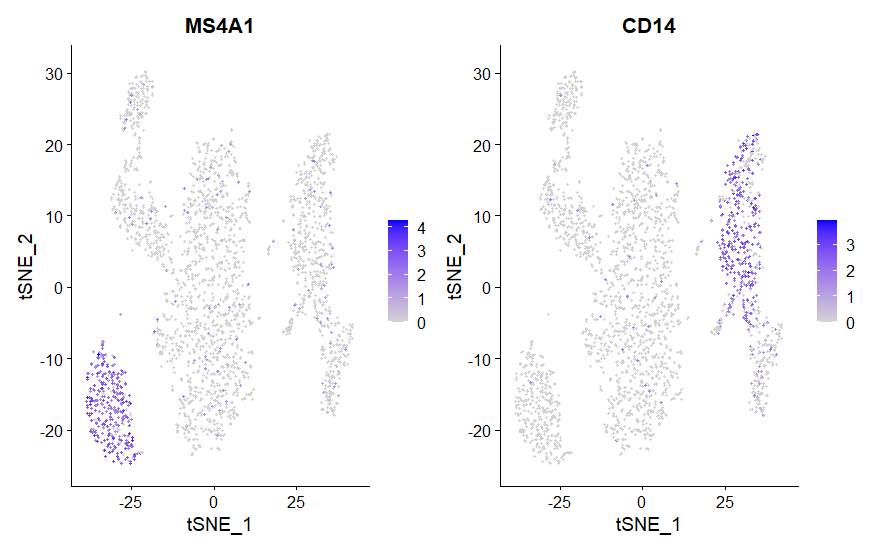
#绘制Marker基因的tsne图;
FeaturePlot(pbmc,features = c("MS4A1","CD14"))
10.寻找亚群的marker基因
10.1 解释标准或参数
Seurat可以找到通过差异表达式定义集群的标记。默认情况下,它识别单个簇的阳性和阴性标记(在ident1中指定),与所有其他细胞相比较。findallmarker为所有集群自动化这个过程,但是您也可以测试集群组之间的相互关系,或者测试所有细胞。
- (1) min.pct参数要求在两组单元中至少检测到一个特征;
- (2) thresh.test参数要求一个特性在两组之间有一定的差异(平均);若两个值都设置为0,会非常耗时——因为这将测试大量不太可能具有高度差异性的features。
- (3) 为了加速,可以设置max.cells.per.ident参数,这将向下采样每个标识类,使其单元格数不超过设置的单元格数。虽然通常会有功率的损失,速度的增长可能是显著的,最高度差异表达的特征可能仍然会上升到顶部。
10.2 举例
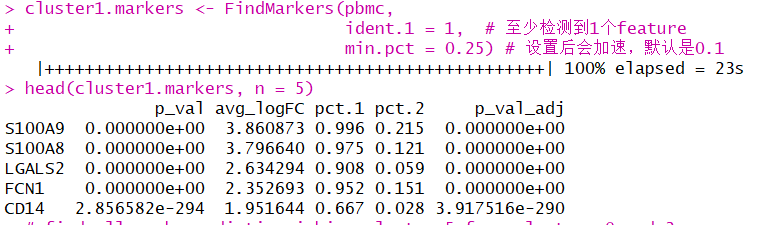
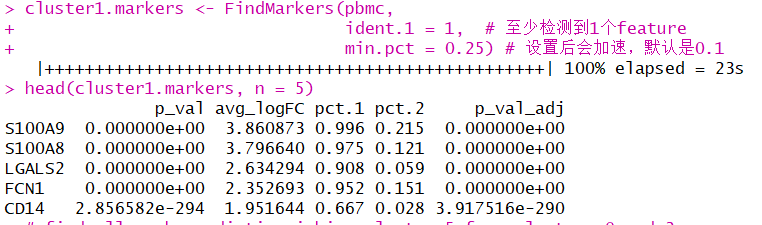
# 找cluster 1的markers
cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, # 至少检测到1个feature
min.pct = 0.25) # 设置后会加速,默认是0.1
# 耗时23 s
head(cluster1.markers, n = 5) # 显示!
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, # 找cluster5的marker
ident.2 = c(0, 3), # 和cluster 0-3对比
min.pct = 0.25)
head(cluster5.markers, n = 5)
# find markers for every cluster compared to all remaining cells,
# report only the positive ones
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25)
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)

10.3 更加精细的寻找marker的方法
我们可以用 test.use 参数设置多种检验方式。例如,用 ROC 返回任何单个标记的“分类能力”。
# 用ROC方法
cluster1.markers <- FindMarkers(pbmc, ident.1 = 0,
logfc.threshold = 0.25,
test.use = "roc",
only.pos = TRUE)
10.4 小提琴图可视化marker
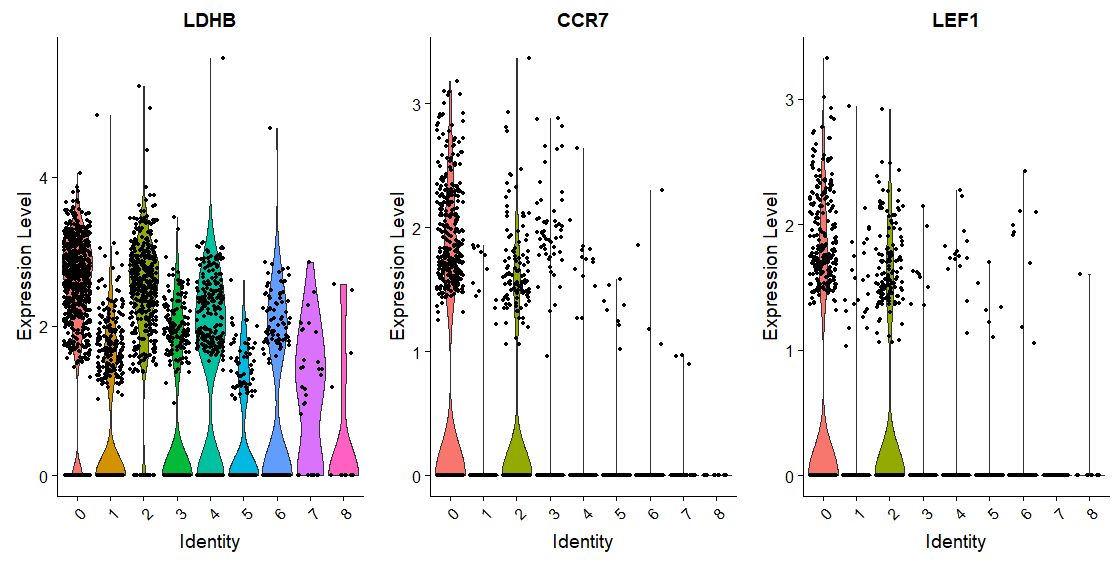
画法1
# 小提琴图将marker可视化,随便挑上部分结果中的3个基因
# LDHB, CCR7,LEF1可以将cluster0分别于其他cluster
VlnPlot(pbmc, features = c("LDHB", "CCR7","LEF1"))
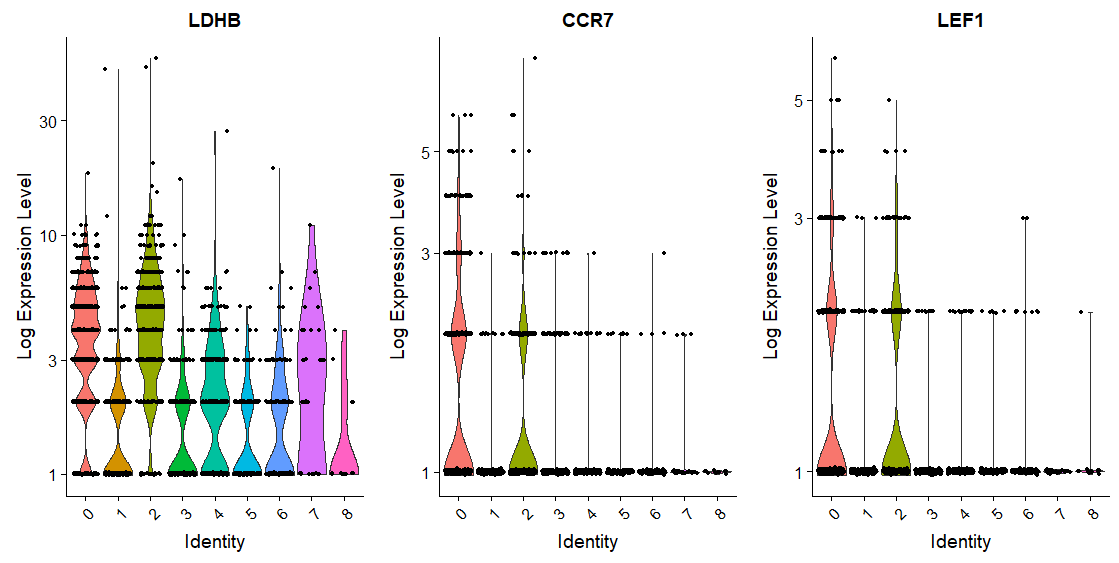
画法2
# raw counts也可以画
VlnPlot(pbmc, features = c("LDHB", "CCR7","LEF1"),
slot = "counts",
log = TRUE)
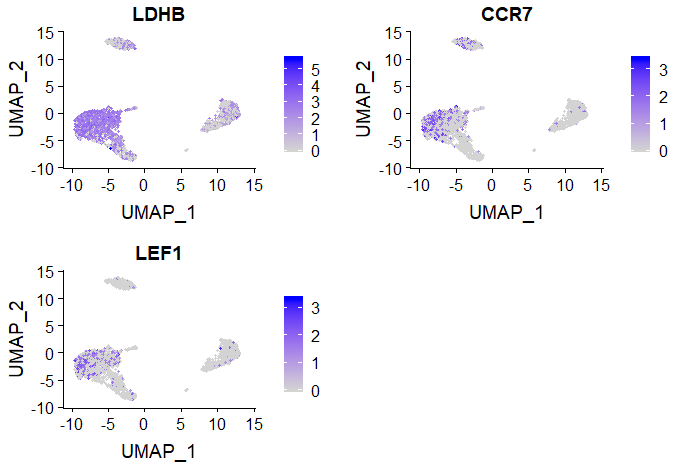
画法3
# cluster 图展示
FeaturePlot(pbmc,features = c("LDHB", "CCR7","LEF1"))
画法4
DoHeatmap为给定的细胞和特征生成一个表达式heatmap。在本例中,绘制每个集群的前20个标记(如果小于20,则绘制所有标记)。
top10 <- pbmc.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_logFC)
DoHeatmap(pbmc, features = top10$gene) + NoLegend()

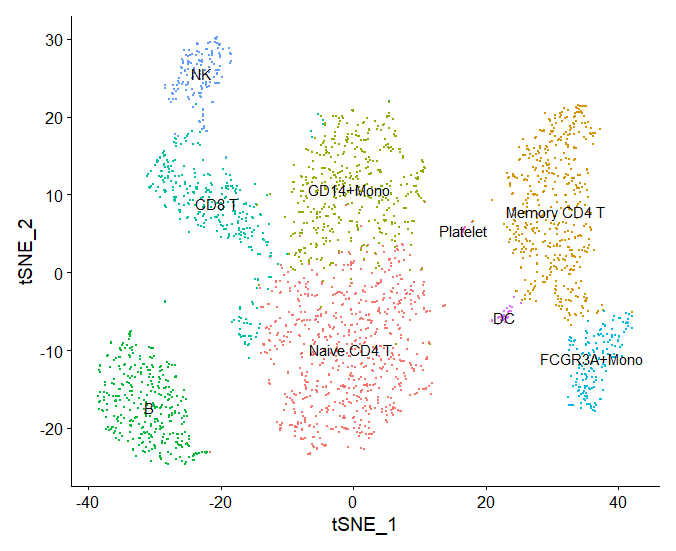
11.为分群重新指定细胞类型
Fortunately in the case of this dataset, we can use canonical markers to easily match the unbiased clustering to known cell types:
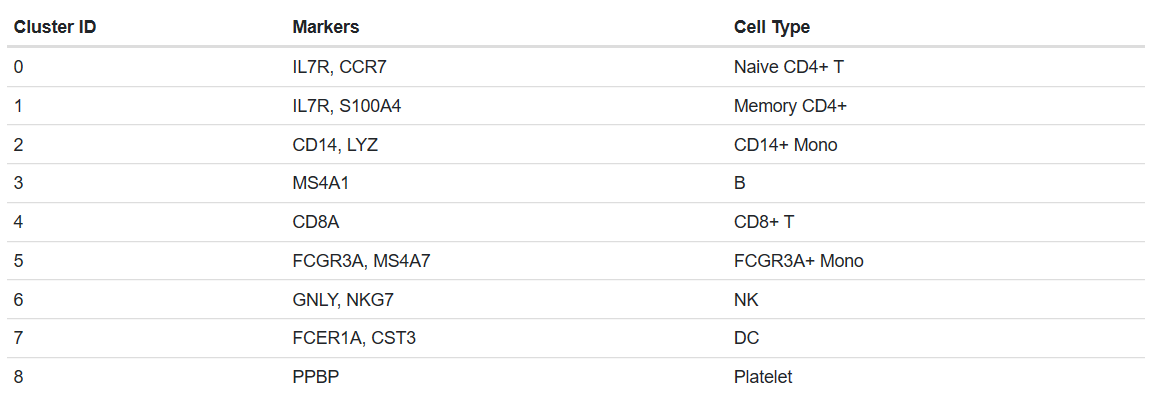
new.cluster.ids <- c("Naive CD4 T","Memory CD4 T","CD14+Mono",
"B","CD8 T","FCGR3A+Mono","NK","DC","Platelet")
#将pbmc的水平属性赋值给new.cluster.ids的names属性;
names(new.cluster.ids)
levels(pbmc)
names(new.cluster.ids) <- levels(pbmc)
names(new.cluster.ids)
pbmc <- RenameIdents(pbmc,new.cluster.ids)
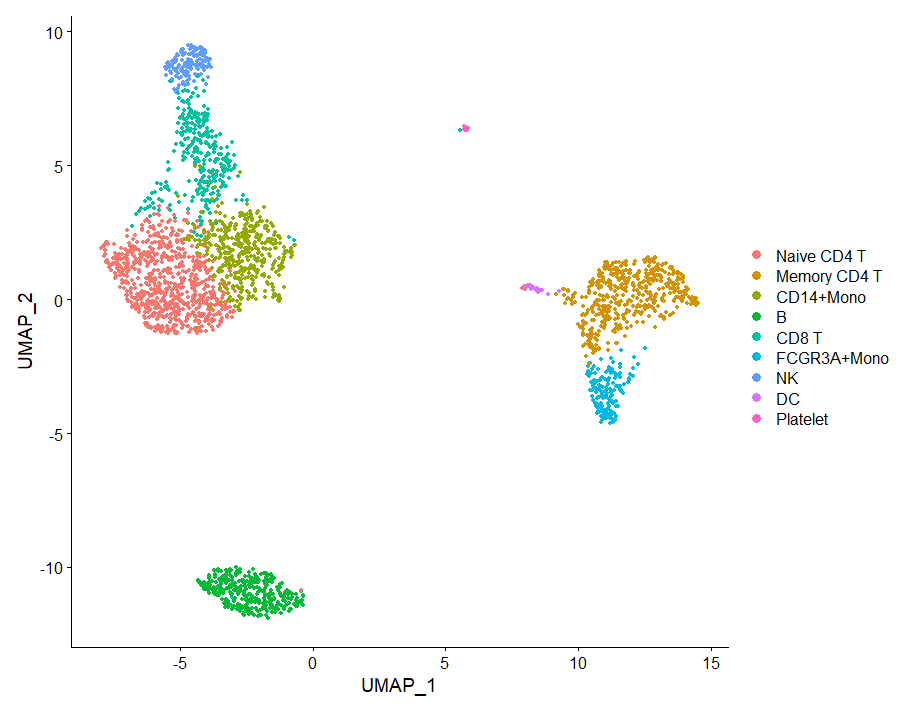
#绘制tsne图和UMAP图(修改标签后的)
tsneplot2 <- TSNEPlot(pbmc,label=TRUE,
pt.size=0.5)+NoLegend()
tsneplot2
uampplot2 <- DimPlot(pbmc, reduction = "umap",
label = TRUE,
pt.size = 1.2,
label.size=5) + NoLegend()
uampplot2