
文章信息:Genetic components of human pain sensitivity: a protocol for a genome-wide association study of experimental pain in healthy volunteers
1.摘要
前言 疼痛是全球疾病负担的主要组成部分。最近的研究表明,遗传对疼痛的敏感性和严重性具有重要作用。尽管大多数可用的证据依赖于候选基因关联或连锁研究,但利用全基因组关联研究(GWAS)对疼痛敏感性的遗传基础的研究仍处于起步阶段。该方案描述了一个关于遗传因素对无关拉丁美洲混合血统的健康个体的基线疼痛敏感性和伤害性敏感性的GWAS。
方法和分析 将在无关混合血统的健康个体中进行一项关于遗传因素对自然状态下疼痛敏感性和伤害性敏感性的GWAS。机械和热疼痛敏感性将通过一系列定量感觉测试来评估疼痛阈值。此外,将评估施用芥末油后局部皮肤的机械和热敏感性的变化。
伦理与传播 这项研究得到了伦敦大学学院研究伦理委员会(3352/001)和安蒂奥基亚大学牙科学院生物伦理委员会(CONCEPTO 01–2013)的伦理批准。研究结果将通过国际会议上的论文和报告分发给委员、临床医生和服务使用者。
2.这项研究的优缺点
-
我们提出了一项关于基线疼痛敏感性以及伤害敏感性的全基因组关联研究。
-
这项研究将在哥伦比亚的混合人群中进行,他们的欧洲、美洲土著和撒哈拉以南非洲血统比例各不相同。
-
表型数据将由一名训练有素的审查员收集,从而产生高质量的测量数据。
-
因为我们专注于在单个中心获得高度可重复的疼痛表型,所以初始样本量仅限于1500-2000名参与者。这将使我们仅识别具有中等和大(但不小的)效应大小的遗传变异。
3.前言
疼痛是全球疾病负担的主要组成部分,腰背和颈部疼痛是影响残障人士生活的主要原因,紧随其后的是偏头痛和其他肌肉骨骼疾病。疼痛是一种多维体验,涉及物理、生化、生理、认知、情感、行为和社会文化因素的高度复杂的相互作用。许多研究已经确定了一系列慢性疼痛状况中的遗传因素。重要的是,有越来越多针对患者人群的研究表明,遗传因素是导致疼痛敏感性和严重性的重要因素。值得注意的是,使用临床疼痛结果或实验性疼痛模型的双生子研究表明,疼痛敏感性的遗传率高达55%。有趣的是,疼痛敏感性的遗传力数值,会随着临床疼痛结果、实验性疼痛模型测试的感觉方式的变化而发生很大差异。有证据表明,不同的感觉模式可能具有不同的遗传组成,从而导致人类在疼痛感知方面的差异,这与强调不同神经生物学介导不同感觉方式的动物模型是一致的。
全基因组关联研究(GWAS)是鉴定人类复杂表型遗传决定因素的一项强有力的技术。然而,GWAS对疼痛的分析仍然有限,主要是因为需要大量的人群样本才能获得足够的功效,以及最终代表个人主观感受的准确表型性状的复杂性。与疼痛变异可能受疾病过程或治疗严重程度影响的疾病队列相比,测量基线疼痛敏感性的实验性疼痛研究具有在标准条件下研究单一刺激的优势(例如,控制强度、位置和刺激持续时间)。到目前为止,已经进行了几项候选基因研究,用实验性疼痛模型来确定基因对人类疼痛敏感性的影响,但是据我们所知,尚未报告完整的GWAS研究。一项全基因组的研究评估了在一组双胞胎中单核苷酸多态性(SNPs)的关联性,这些双胞胎的外显子组测序被认为具有特别高或低的热痛敏感性。通路分析表明,血管紧张素通路中的基因变体出现了显著性富集。然而,实验性疼痛敏感性和临床疼痛严重程度之间通常不存在直接联系,有一些证据表明,实验性疼痛模型的发现可以预测临床相关的病理性疼痛,如术后疼痛。无论实验性疼痛和病理性疼痛之间有何关联,了解基因对实验性疼痛敏感性的影响将为疼痛敏感性的机制提供重要的生物学见解。
迄今为止,疼痛领域中大多数遗传研究的一个重要缺点是,这些研究主要在欧洲祖先群体中进行,因此,他们只研究了人类表型和遗传多样性的一小部分。这一点在疼痛的研究中很重要,因为最近有研究指出,欧洲裔美国人、非裔美国人和拉丁裔的疼痛阈值具有差异性。然而,目前尚不清楚疼痛阈值的变化是否与神经生物学机制的差异或其他因素(例如社会或文化因素)有关。此外,最近的研究表明,有一类表型与时间累加引起的伤害感受系统的敏感性相关,这可能使人们容易产生临床相关的疼痛。然而,大多数研究集中在自然状态而不是敏感状态下的疼痛表型。只有少数研究研究了伤害性敏感的遗传组分。因此,我们打算使用一种致痛剂(异硫氰酸烯丙酯[AITC],配体门控离子通道TRPA1的激动剂)来敏化伤害感受系统,以复制病理性疼痛状态下可能发生的变化。
该方案的针对人群是与欧洲/美洲印第安人/非洲人血统无关的健康个体,并试图通过GWAS来研究基线疼痛敏感性和伤害敏感性的遗传贡献。我们将评估基线皮肤疼痛阈值以及芥末油(AITC)施用后的敏感性变化,芥末油是一种组织损伤的受控模型。我们将确定与自然和敏感状态下的实验性疼痛刺激相关的SNPs。阐明疼痛变化的遗传基础,有可能为今后镇痛药物的开发提供靶标。这一研究成果还可以根据不同人群之间的敏感性差异来改善疼痛管理方案(针对个人的疼痛风险或恢复力进行调整)。
4.材料和分析
该GWAS遵循《加强遗传协会研究报告的指南》。研究程序流程图如图1所示。
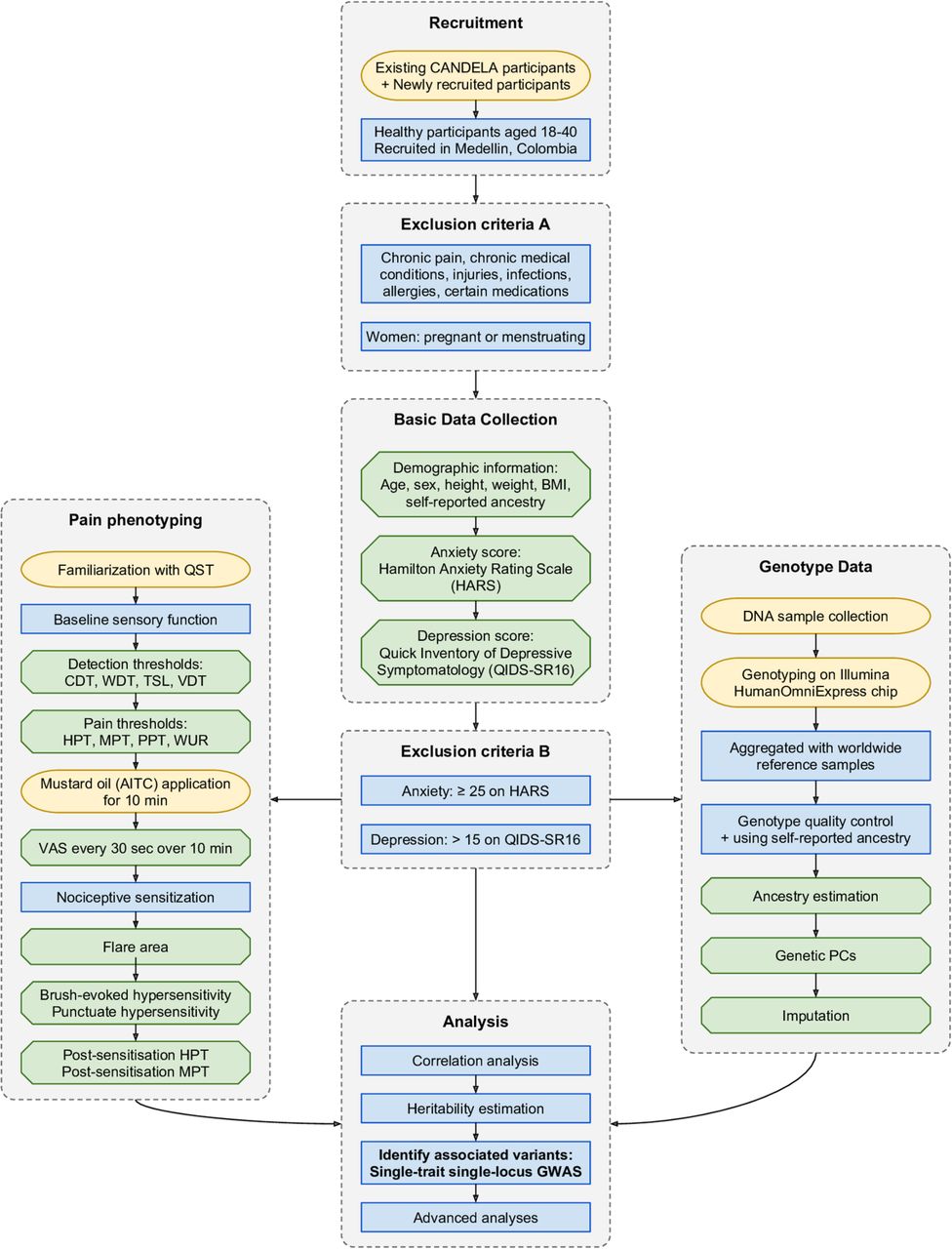
图1研究过程。
研究程序。图表详细说明了从招聘到数据分析的研究过程。黄色框表示新的过程;绿色框表示数据生成和收集;蓝色框表示过程步骤。AITC,异硫氰酸烯丙酯;BMI,体重指数;CANDELA,拉丁美洲多样性和进化分析协会;CDT,冷检测阈值;GWAS,全基因组关联研究;HPT,热痛阈;MPT,机械痛阈;PCs,主成分;PPT,压力痛阈;QST,定量感官测试;TSL,热感阈值;VDT,振动检测阈值;VAS,可视化模拟量表;WDT,温感阈值;WUR,缠绕比。
4.1 参与者
18-40岁的健康参与者将在哥伦比亚麦德林通过当地大学的公共布告栏、传单分发和当地印刷媒体招募。此外,我们还邀请了先前参与GWAS研究的CANDELA志愿者来参与本项目。
招募健康的年轻参与者在GWAS研究中具有优势。他们不太可能有未被发现的疾病或其他可能影响其疼痛敏感性生物学途径的问题。年轻人的总体累积暴露量和风险较少,这些外部环境因素可能会影响他们对疼痛的敏感性。 这些因素增加了参与者疼痛感知反应的整体可变性,降低了检测遗传因素的功效。由于大多数性状都受到遗传和环境因素的共同影响,包括CANDELA在内的许多研究都倾向于使用年轻的参与者来进行遗传变异的鉴定。
患有慢性疼痛或任何慢性医学疾病(例如糖尿病、神经退行性疾病、肌肉骨骼疾病或精神疾病)的参与者将被排除在研究之外。目前正在服用止痛药、抗炎药、阿片类药物、抗组胺药、抗抑郁药或抗癫痫药的参与者将被排除在研究之外。怀孕或处于月经期的妇女将被排除在研究之外。我们建议参与者在测试前1小时内不要吸烟或喝咖啡,并在测试前8小时内避免使用神经活性物质或酒精。进一步的排除标准包括:当前或过去的自残伤害,影响手臂的皮肤病、创伤或感染性疾病,以及对任何药物、材料、食物或昆虫叮咬的严重过敏反应史。中度至重度焦虑(汉密尔顿焦虑评分量表≥25)或重度抑郁(QIDS-SR16>15)的受试者被排除在研究范围之外。招募工作于2013年1月开始,预计需要大约5-7年的时间。
4.2 程序
参与者将参加麦德林安蒂奥基亚大学定量感官测试(QST)实验室的一次会面。在知情同意后,将记录年龄和自我报告的性别,参与者将回答有关他们自我报告的祖先的问题。将测量参与者的身高和体重,并计算他们的体重指数(BMI)。由于焦虑等心理因素会影响实验性疼痛测试过程中的疼痛感知,因此参与者将完成西班牙语版的汉密尔顿焦虑量表和QIDS-SR16。与其他抑郁评分相比,QIDS-SR16具有可接受的内部一致性和中至强的并发效度,其西班牙语版显示了足够的测试-复测信度和高的内部一致性。汉密尔顿焦虑评分表显示出较高的评分和测试-复测可靠性,并且具有良好的结构有效性。
4.3 评估自然状态下的感觉功能
我们将确定自然状态和伤害性敏感下的感觉功能。将使用特定的静态和动态QST来评估基线感觉功能。其中包括冷检测阈值(CDT)和热检测阈值(WDT)的检测,以及使用ThermoTester来检测热感觉阈值(TSL)和热痛阈值(HPT)。热阈值的记录将严格遵循已发布的QST指南。机械疼痛阈值(MPT)将使用20根Frey hair set,通过施加不同力度的方式进行评估(9.8, 13.7, 19.6, 39.2, 58.8, 78.5, 98.1, 147.1, 255.0, 588.4, 980.7, 1765.2, 2942.0 mN)。从9.8mN基线刺激开始,将以2s开2s关的速率升序施用von Frey hairs,直到参与者感觉到尖锐刺激(产生刺破感)。随后,将hairs的力度降序施用于受试者,直到刺激被视为钝感为止。将五组升序和降序力度刺激的几何平均值定义为MPT。Wind-up ratio(WUR)将通过可视化模拟量表(VAS 0–100)上单个刺激的数字疼痛评级来确定,然后使用255mN的von Frey hair,在相同的1cm^2范围内,以1Hz的频率对10个刺激进行平均疼痛评级。这将被重复五次,并且该比率将被确定为一系列刺激的平均等级除以单个刺激的平均等级。振动检测阈值(VDT)将通过使用Rydel-Seiffer音叉记录3个消失阈值的平均值来确定。使用手动重力计记录压力疼痛阈值(PPT),一式三份,并将其平均值用于分析。
将随机分配受试侧手臂。患者首先应在对照侧的前臂熟悉前臂的感官测试,然后再对测试臂进行实际测量。除VDT(ulnar styloid)和PPT(thenar muscles)外,所有测试均前臂掌侧的一半区域进行。
4.4 芥末油诱发的伤害性敏感
在基线感觉测量之后,将使用醋酸纤维模板标记一个有八个辐条的星形,每个辐条在前臂掌侧以1cm的增量包含八个点(图2)。在开始之前,将32℃的温热电极放置在恒星中心5分钟,使皮肤温度标准化。然后,我们将使用芥末油(AITC(Sigma),在橄榄油中稀释30%)进行敏化试验。芥末油的活性成分AITC能够激活离子通道TRPA1,并引起皮肤红晕和伤害性敏感。将一块浸有芥末油的小棉签涂在掌前臂上0.64cm^2的面积上,并用Tegaderm(3M)固定10分钟。在此期间,每30s使用0到100的电子VAS记录疼痛评分。10分钟后,去除芥末油,并记录每个辐条处皮肤红晕的面积,精确到0.5cm。通过将相邻辐条上的点连接起来,可以创建八个三角形,并通过添加所有三角形线段来计算总面积。从总面积中减去施用芥末油的面积,即可确定次生红晕的面积。
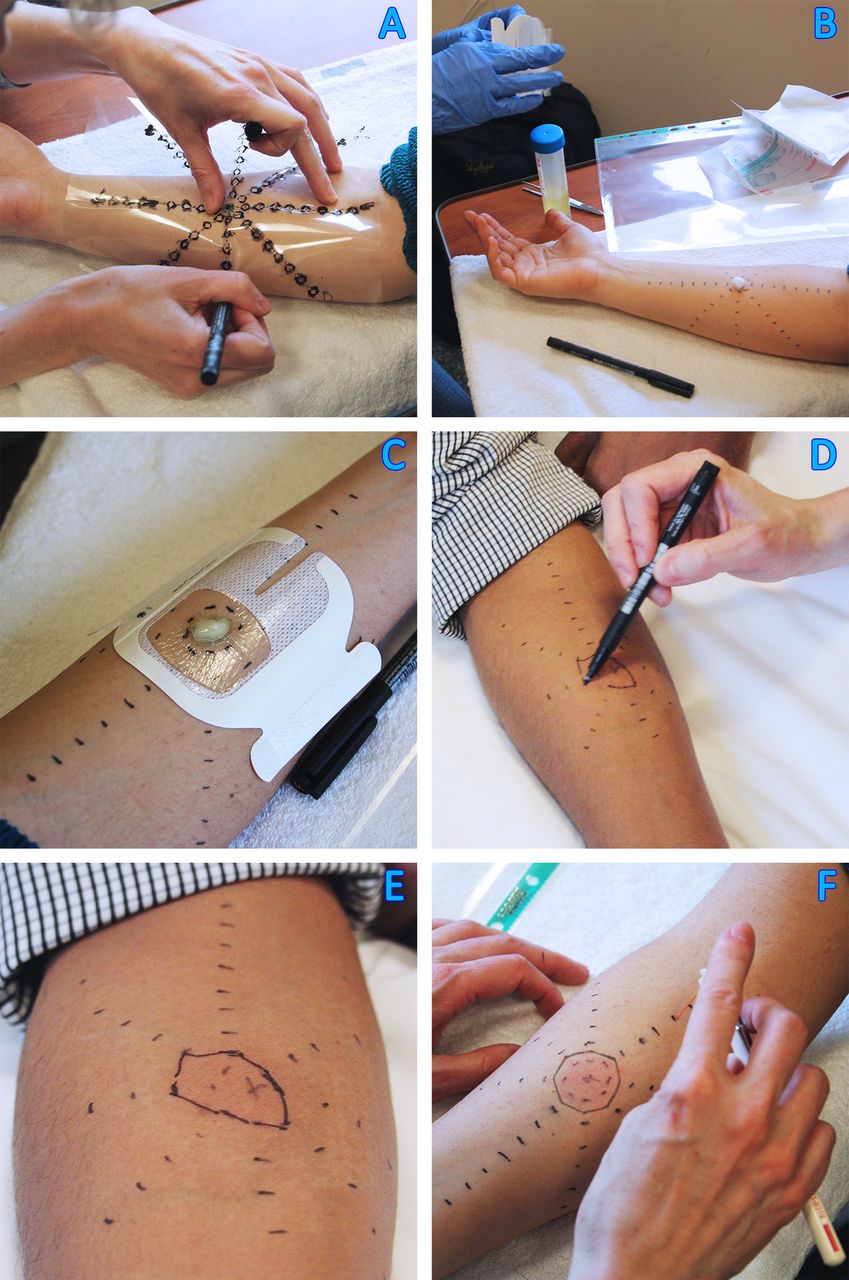
图2 确定红晕、点状痛觉过敏和异常性疼痛的方法
(A)醋酸模板用于绘制星形标记,其中八个辐条包含8个点,增量为1cm。
(B)在星形标记的中央涂抹浸有30%芥末油的小棉签。
(C)用Tegaderm固定10分钟。在此期间,每30秒记录一次疼痛评分。
(D和E)去除芥末油后,将标记出皮肤红晕并计算出面积。
(F)通过测试八个辐条上每个点的潜在敏感性,来确定brush诱发的过敏区域以及von Frey hair诱发的点状过敏区域。
绘制好红晕区域后,将使用brush在八根辐条上的每个点上施加1厘米长的笔触,从外侧开始,向敏感中心移动,以确定brush诱发的敏感区域。按照相同步骤,用98.1mN的灯丝测定点状敏感的区域。至于红晕,将从两个过敏区域中减去芥末油施用的主要区域,以使记录的区域代表次生(secondary)痛觉过敏/过敏症。
芥末油敏化后,将使用上述相同方法重复MPT和HPT。所有的敏化后测试将在芥末油去除后5分钟内进行。
4.5 自然和敏感状态下的感觉功能方案的可靠性
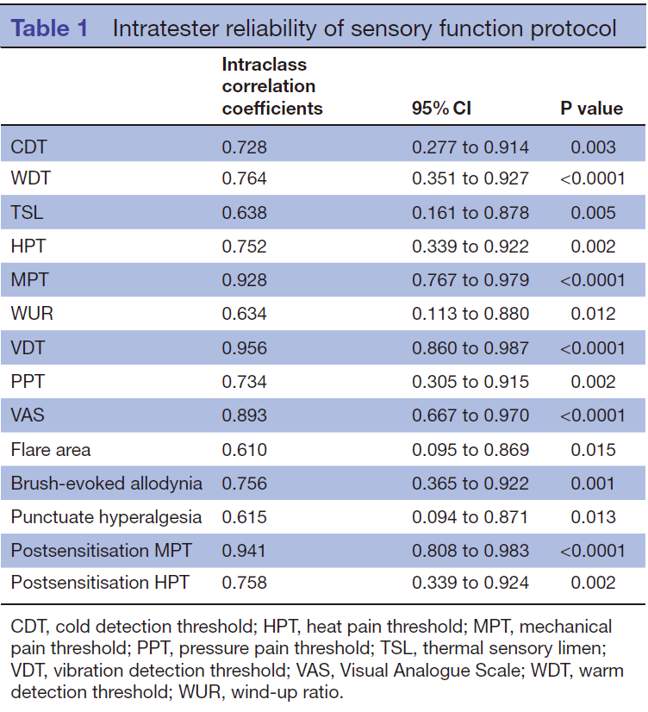
为了确定内部测试的可靠性,我们在2-6周内的两个不同场合重复了同一研究者在n=12名健康志愿者中进行的感觉功能方案。组内相关系数(3.1)显示,所有感觉测试变量的一致性为良好至极好(表1)。
表1 感觉功能方案的测试人员可靠性
4.6 基因分型
每位参与者将捐献血液或唾液以用于DNA提取。DNA样本将在Illumina HumanOmniExpress芯片上进行基因分型,该芯片包含约700000个标记。在已经参与CANDELA GWAS的志愿者中,来自同一芯片上基因型的血液样本的39个基因型数据已经可用,并将被重复使用。
我们将对Illumina阵列的全基因组基因型数据进行质量控制,以排除任何不符合严格阈值的标记或样本。Illumina GenomeStudio软件中基因型调用算法提供的质量指标(如GenTrain评分、聚类分离评分和过量杂合率)将用于过滤基因型较差的SNP。随后将应用SNP水平和sample水平的质量控制阈值(例如缺失)。我们将检查X和Y染色体的记录和遗传数据之间的性别错配。能够通过上述所有标准的sample和SNP将被保留用于后续分析。
4.7 统计分析
4.7.1 样本大小评估
根据Visscher等人描述的公式,对不同样本和效应大小的实验性疼痛表型的GWA的功效进行了估计。从现有实验性疼痛研究中获得的实验性疼痛表型的一系列效应大小显示了估计的功效。使用R上的统计软件TEam RC.R进行计算并生成图表。代码发布在https://github.com/kaustubhad/gwas-power。
在基于全基因组SNP的GWAS研究中,通常采用多元线性回归模型进行关联分析,表型值最终会回归到SNP基因型和其他协变量(通常包括年龄、性别、BMI和遗传主成分(PCs))。全基因组显著关联的p值阈值通常为5×10^-8,而边际(suggestive)显著关联的阈值通常为10^-5。在线补充附录3中提供了用于计算GWAS中具有全基因组范围和边际显著性阈值功效的公式,图3显示了针对当前GWAS设置计算的功效。
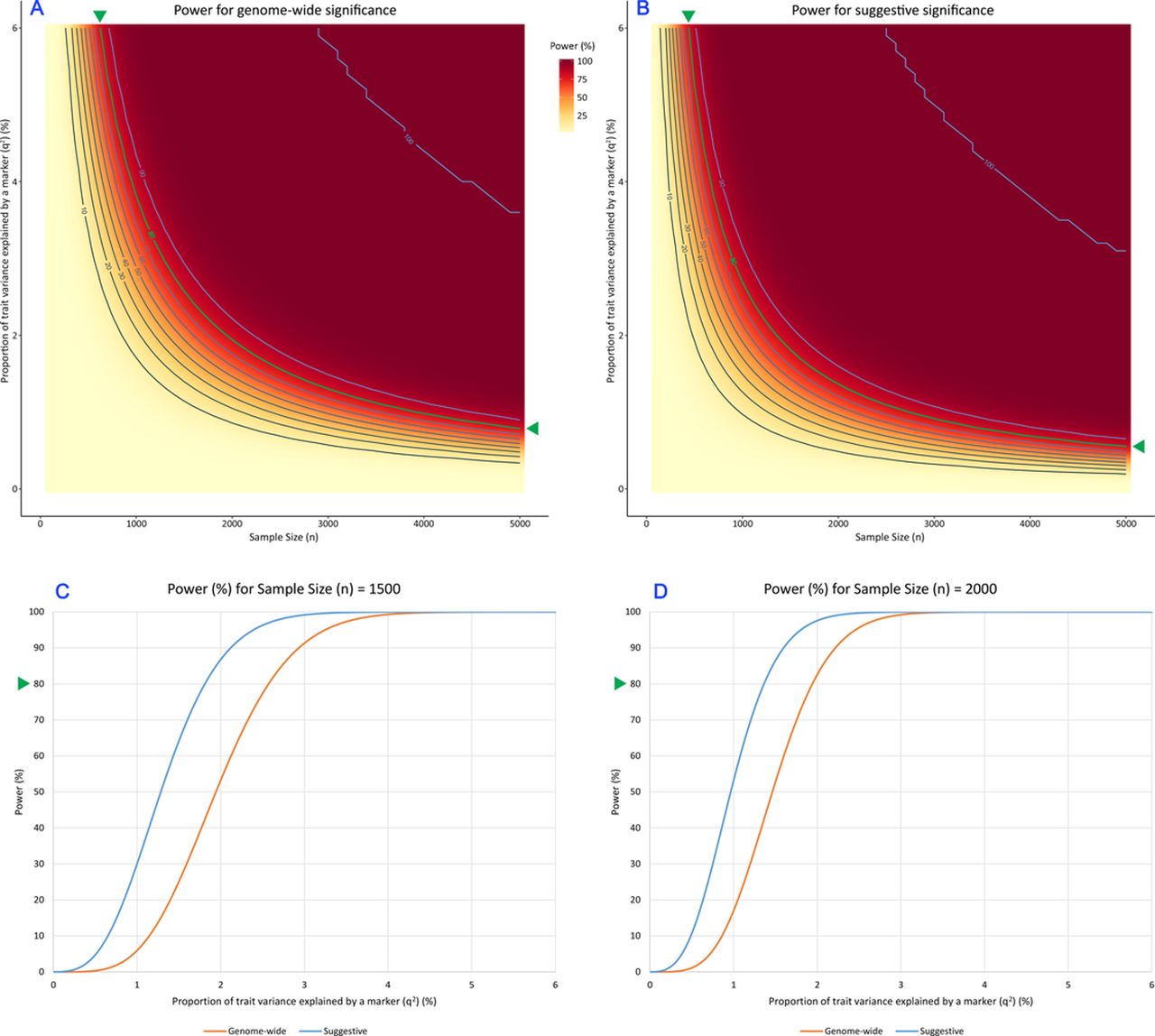
图3 在使用全基因组基因分型数据的标准全基因组关联研究(GWAS)设置下的估计功效(以百分比表示)。
(A)将估计功效(以百分比表示)作为热图,将显著性阈值设置为5×10^-8,这是GWAS研究中普遍使用的全基因组显著性阈值。
(B)将显著性阈值设置为10^-5(边际显著性的常用阈值)的估计功效。在A-B中,x轴表示GWAS中样本大小(n)的范围,y轴表示方差解释度(q2)。颜色梯度表示在特定(n,q2)组合下检测SNP的能力。还显示了10%间隔的功效等值线。C-D显示了本研究的预期样本量的功效曲线。
(C)样本量n=1500的全基因组和边际显著性阈值的预期功效。
(D)样本量n=2000的估计功效。
在C-D中,x轴表示SNP对表型的方差解释度(q2),y轴表示估计的功效(百分比)。两条曲线对应于两个常用的GWAS阈值。在每个图中,用绿色三角形表示80%的功效点,以便可以从图形中读取必要的参数配置。在A-B中,对应于80%功效的轮廓也用绿色标记。
图3A显示了在标准GWAS设置下使用全基因组基因分型数据和p值显著性阈值为5×10^-8的估计功效(以百分比表示)作为热图。样本量(n)在100到5000之间变化,而SNP对表型的方差解释度(q2)在0.01%到6%之间变化。随着样本量的增加,一系列表型方差值达到100%的功效迅速增加。
图3B显示了相同设置下的估计功效(以百分比表示),但边际p值显著性阈值为10^-5。不出所料,对于这个不太严格的阈值,在相似的样本和效应大小下,功效更高。
本研究预期样本量的简化功效估算如图3C、D中的功率曲线所示。图3C显示了在不同效应大小下,n=1500样本量在全基因组和边际显著性阈值下的预期功效,而图3D显示了n=2000样本量的估计功效。
表型方差的范围取自Doehring等,它提供了几个实验性疼痛表型和多个SNP对表型的方差解释度的估计值。值范围为0.02%至6%。有些表型与本文研究的表型相同,而有些表型则不同。然而,两组表型的表型方差分布非常相似,如图4A所示。
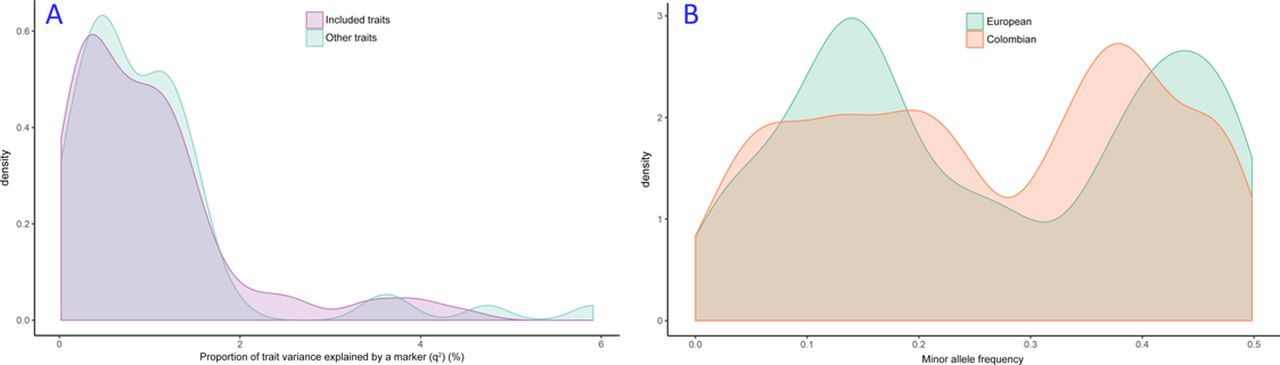
图4
(A)Doehring等人的一个SNP解释了我们研究中包括和未包括的表型的表型方差分布。
(B)先前发表的针对欧洲人和哥伦比亚人队列中与实验性疼痛相关的基因座的等位基因频率分布。
GWAS的功效取决于SNP的等位基因频率对测试统计量的影响。尽管大多数GWAS研究是在欧洲人中进行的,包括用于确定样本量的实验性疼痛研究,但我们感兴趣的人群是拉丁美洲的混合人群。因此,我们想评估在各种研究中针对实验性疼痛或与之相关的SNP在欧洲人和哥伦比亚人中等位基因频率的分布。我们从1000人基因组项目数据库中获得了西欧人(来自英国(GBR)、北欧和西欧犹他州居民(CEU)、西班牙(IBS)和意大利托斯卡纳人(TSI))和哥伦比亚人(来自哥伦比亚麦德林(CLM),本研究将在这里进行)的所有已公布的SNP次要等位基因的频率。欧洲人和哥伦比亚人的等位基因频率分布如图4B所示。两种分布非常相似,而哥伦比亚的分布稍微分散一些。由于哥伦比亚人平均拥有60%的欧洲(西班牙)血统,这在一定程度上是可以预期的。在GWAS中,等位基因频率的良好分布很重要,因为对于给定的样本和效应大小,低频等位基因的功效较低。在这里,与欧洲等位基因频率分布的比较表明,当前的哥伦比亚队列将具有与欧洲人队列相同的功效。与仅包含欧洲人的队列相比,我们的队列将有一个优势,即在其他大陆人群中存在的等位基因,如撒哈拉以南非洲人或美洲土著人中不存在的等位基因,也可以在GWAS中检测到,并且可以在特定种族的复制队列中进行随访。
CANDELA项目包括约2000名麦德林患者的基因型。我们期望与其中50%–75%的参与者进行联系并对他们的表型进行采集;此外,我们还会对另外500名参与者进行进行表型采集,目的是使得初始样本量达到1500–2000。
4.7.2 数据分析计划
清洗后的遗传数据将首先与全球参考样本合并,例如1000人基因组计划、Simons基因组多样性计划、爱沙尼亚生物中心人类基因组多样性小组以及与拉丁美洲人口特别相关的其他欧洲人和美洲印第安人样本。我们将通过遗传PC和大陆血统比例(使用Admixture软件)检查合并后的数据集的遗传异常值,以及是否存在意料之外的遗传相似性。这些步骤通常可以检测到任何样本错位或污染,这可能反映在性别错配、意料之外的遗传相似性或过高的杂合率上。遗传祖先估计值将与自我报告的祖先信息进行比较,特别是对于遗传异常值或显示出乎意料结果的样本。对于哥伦比亚人中罕见的种族,如东亚或南亚,参与者的自我报告也将被排除在外。作者在混合人群中进行关联分析方面具有丰富的经验,已发表许多涉及到一系列表型的GWAS文章,其中包含有关如何进行此类分析的详细方案。
遗传数据可以估计任何数量性状的狭义遗传力,即由遗传数据解释的表型方差的分数。使用GCTA软件获得的遗传力估计值将提供一个思路,即哪些表型更具有生物学基础,而哪些表型更受环境影响。上述思路将提醒我们哪些表型更适合进行遗传分析以发现相关的遗传变异。但是请注意,通过这种方法得出的相对精确的遗传力估计(低SDs)需要数千个样本,因此本文中提出的样本量可能无法准确估计遗传力。
为了更好地识别相关基因座,基因型数据将使用1000 Genomes phase 3 imputation reference panel,首先使用SHAPEIT2进行第一次单倍型定相,然后使用Impute2进行插补,从而插补到大约1000万个基因座。将使用推荐的插补质量分数阈值、一致性度量和高概率调用比例对插补基因型进行质量控制。
GWAS研究将在Plink2中进行,以在加性多元线性回归模型中对整个基因组中的每个表型单独进行单SNP关联研究。协变量将在回归中用于调整表型变异的任何其他来源,如年龄、性别和体重指数等基本变量,遗传PC将用于控制群体亚结构
回归中包含的遗传PC数取决于样本组成,例如祖先的变异和是否存在遗传异常值。可以通过检查每个PC的方差解释度(显示在碎石图上)并检查PC散布图来确定。
除了作为排除标准外,焦虑和抑郁得分也可以作为GWAS的协变量。将根据初始诊断分析(如相关分析)确定要使用的确切协变量集。
这些作为p值获得的SNP关联结果将通过曼哈顿图可视化。选择相关基因座的常用p值阈值为5×10^−8(全基因组显著性)和10^-5(边际显著性)。
GCTA还将对仍在单表型单SNP设置中的这种加性多元线性回归模型进行扩展,称为混合线性模型分析,该模型可以更好地控制任何隐秘的相关性或总体子结构。
单表型单SNP关联研究有几个扩展,可以提高检测相关基因座的能力:使用MultiPhen中实施的多变量Wald检验,结合可能共享生物学基础的几个相关表型;或基于基因的测试,将基因中所有位点的信号组合起来,以提高信号强度并减轻多重测试的负担,例如Plink2中实现的基于集合的模型或GCTA中实现的fastBAT。样本的混合性质可通过混合作图法检测关联,尽管这种方法在检测相关变体方面的成功可能性取决于变体等位基因频率在各大陆的分层程度。这些分析可能有助于检测由于较小的效应大小而在经典GWAS中动力不足的额外SNP位点。
4.7.3 缺失数据处理
即使前100个样本的完整性表明缺失率很低,也可能无法在某些个体中记录某些表型。传统GWAS分析中使用的单表型方法会自动从表型分析中排除那些缺少该表型值的个人。对于任何特定SNP具有缺失基因型的个体也是如此。但是,在CANDELA队列中使用Illumina HumanOmniExpress芯片进行基因分型的成功率非常高(>99.8%),因此,任何分析中排除个体的数量总体上将非常低。
一些多变量分析(例如应用于表型的PC)要求记录个人样本的完整表型值。与其使用记录了完整表型的个体子集(这会造成样本量的损失),不如按照R包“mice”中实施的标准统计程序估算每个个体的缺失表型数据。当丢失数据的比例较小时,在这种多变量分析中,插补比样本排除更可取,并且在计算遗传PC时通常将其应用于遗传学数据。
5.讨论
该GWAS项目包括一个定义明确的健康参与者队列,将为在自然和敏感状态下实验性疼痛敏感性的遗传基础提供重要的见解。这可能允许进一步探索潜在的疼痛敏感性生物学机制。未来还需要进行进一步的研究以将这些发现推广到患有慢性疼痛的患者人群中。
6.专利和公众参与
在这项研究中没有患者参与。
7.伦理与传播
研究结果将通过论文和在国际会议(例如两年一度的国际疼痛研究协会世界大会)上的演示文稿分发给专员、临床医生和服务使用者。我们还将把研究结果发布到可公开获得的painnetworks.org数据库中。
8.致谢
略。
9.参考文献
略。