1.什么是单细胞测序
普通测序所提取的RNA(或DNA)源于样本中的多个细胞,所以普通测序的结果不可避免的会受到不同细胞间异质性(Heterogeneity)的影响,而单细胞测序(Single-cell sequencing)则是针对单个细胞的基因组进行测序,能够更好地帮助我们认识细胞与细胞之间的差异。单细胞测序首先不是仅仅对一个细胞进行测序,而是说该项技术能对单一细胞的基因组或转录组进行测序,可以理解为单细胞水平上的测序。
2.单细胞组学研究的特点(相比于常规组学)
- (1)包含多种组学,自成体系:RNA-seq、ATAC-seq、DNA-seq等;
- (2)分析更加复杂:数据规模和复杂成都扩大了三四个数量级;
- (3)样本准备要求更高;
- (4)技术发展日新月异:要求科研人员不断学习,不断提高。
3.单细胞测序的发展由来(为了解决什么问题)
- (1)细小组织的划分问题(研究目标是更具体更细小的局部组织)
- (2)细胞异质性(常规转录组检测的是样本中所有细胞的均值)
4.单细胞测序的发展史
整体特点是测序通量越来越高(即在单位时间和费用内,可以获得更多单位细胞的信息)、技术稳定性越来越高(从而保证结果稳定性)。
从通量角度来看,单细胞测序可以分为三类技术:
- (1)一次只能检测1个细胞的技术,代表性的技术是Smart-seq2;
- (2)基于微流控芯片等,一个批次可以检测100个左右的细胞,例如 Fluidigm C1;
- (3)一次可以批量检测成千上万个细胞的技术,包括SPLIT-seq、sci-RNA-seq、10X Genomics等。
其中10X Genomics是最具代表性的技术之一,其关键特点是系统稳定性高,目前已被大规模应用。
5.单细胞测序的应用
单细胞测序涉及不同组学,有单细胞基因组测序、单细胞DNA甲基化测序、单细胞ATAC-seq测序,但现在最火的还是单细胞转录组测序。10X Genomics单细胞测序,是这两年来生物科研领域最火的一个技术之一。10X Genomics技术不仅仅拘泥于为转录组检测,还拓展出了单细胞CNV-seq、ATAC-seq、免疫组库检测、Crisper筛选等产品。在基础科学领域,拥有不同组学的产品意味着用户可以围绕10X Genomics技术开展多组学贯穿,获得更丰富的研究思路和解决方案。
6.单细胞转录组的主要步骤及关键点
单细胞捕获/分选 ——> 反转录/PCR扩增 ——> 建库测序 ——> 生物信息分析
其中非常关键的一点就是如何进行单细胞的捕获/分选,这是决定单细胞检测成本和通量的关键步骤。在细胞分选的方法里,主要包括特异性分选(基于特定标志)和非特异性分选(高通量)两类方法。
- (1)特异性选择:微吸管分离、激光捕获显微分离、荧光激活细胞分选(FACS);
- (2)高通量非特异性选择:微流控芯片(Fluidigm C1)、微液滴分选(10X Genomics)、微孔(BD Rhapsody)。
7.10X Genomics平台
相比Fluidigm类技术使用实体芯片作为隔离单个细胞的载体,10X Genomics技术则是使用液滴作为隔离单细胞的载体。10X Genomics产品基于Drop-seq,该技术的思想是在流动的管道中形成油包水的微液滴体系,让每个单细胞在微液滴中完成相关反应。
10X scRNA-seq包括细胞悬液制备(单细胞)、文库构建(包含细胞分选)、测序、数据分析、可视化等步骤。
8.10X Genomics平台中文库构建的步骤
- (1)准备好10X官方提供的微磁珠(每次反应大概投入75万个磁珠,数量远大于投入的细胞数)。这些磁珠上都连接着后续反转录所需的adaptor(接头)、barcode(用于区分细胞)、UMI(用于区分mRNA分子)等序列。
- (2)磁珠进入10X Genomics芯片。磁珠可以在管道中流动(管道中是水),会经过两个“十字路口”。在第一个“十字路口”与另外一个管道交叉。新加入的管道里流动的是细胞和反转录扩增所需的试剂。经过这个“十字路口”后,一部分磁珠就吸附上了细胞,且整个液体环境富含后续反应所需的酶。在第二个“十字路口”,新加入的管道里流动的是油。油和水混合,就会形成乳浊液(油包裹水滴的环境)。由于磁珠是亲水的,所以一个个磁珠以及其吸附的细胞就被包裹在水滴里。我们将这些油包水的微液滴,里面含有磁珠的结构称为GEM(Gel bead in emulsion)。
- (3)在这些GEM里,将完成细胞裂解、反转录、扩增等步骤。
- (4)每个细胞的cDNA完成扩增,并携带这独一无二的接头,等待进行后续测序。
基于这样的步骤,通过合理控制每次输入的细胞数,最终一张10X Genomics芯片可以检测的细胞数一般控制在3000-8000个之间。而且因为检测的细胞数巨大,几乎保证每类细胞至少几十个甚至成百上千个重复。
9.10X Genomics单细胞转录组是测全长还是单端
10X Genomics单细胞转录组并非测mRNA全长,而是通过测mRNA 3’端或者5’端实现定量。这里本质的逻辑就是我们对mRNA进行定量,只需要测定RNA分子的条数。那么测一段和测全长是等效的,显然只测其中的一段是更简单更高性价比的策略。当然,因为只测mRNA分子的3’端或者5’端,自然就无法开展可变剪切分析了。
10.空间转录组测序(Spatial Transcriptome sequencing)
空间转录组是以分析样本内部不同空间位置转录组信息为目的的技术。10X scRNA-seq虽然将转录组精度提高到了细胞水平,但在制作单细胞悬液的时候,每个细胞也丢失了空间信息。即这些细胞来自组织的哪个位置,组织不同位置的基因表达特性如何,这些信息是10X scRNA-seq无法告诉我们的。
| 技术类型 | 检测通量 | 位置信息 | 单个转录组构成 |
|---|---|---|---|
| 10X scRNA-seq | 300-10000个细胞 | 丢失 | 单个细胞 |
| 空间转录组 | 最高5000个位点 | 精确记录 | 1-10个细胞 |
空间转录组的应用价值:
- (1)转录组信息更好与组织形态的特征关联
- (2)可以与10X scRNA-seq技术互补
11.10X单细胞技术是否需要参考基因组
scRNA-seq或者ST-seq,本质上就是转录组,研究的目标分子是带ployA尾巴的RNA。因此,并非必须要有参考基因,只要有质量足够好的参考转录本就可以了。VDJ-seq受限于试剂只针对人和小鼠开发,因此其他物种目前无法开展商业化的服务。ATAC-seq作为检测基因组开放性的技术,其检测的区域大部分为非编码区,因此参考基因组不但必须要有,而且参考基因组的质量对ATAC-seq的影响非常大。
12.10X scRNA-seq/ST-seq可以检测哪些类型的RNA
(1)mRNA:由于真核生物mRNA都有ployA结构,所以理论上mRNA就是10X scRNA-seq/ST-seq主要的检测目标。当然,由于只是扩增mRNA 3’端或者5’端的一小段用于定量,所以并不能用于分析可变剪切。
(2)lncRNA:高等生物的lncRNA只有一部分有ployA结构,因此10X scRNA-seq/ST-seq只能检测这些有ployA结构的lncRNA。另外,由于lncRNA表达量普遍比较低,而10X scRNA-seq/ST-seq这类大规模单细胞/准单细胞测序的技术,对低丰度mRNA分子的检测能力比较弱,因此结果中lncRNA的数量将比较少。
(3)其他RNA:近年来研究大热的环状RNA由于没有ployA结构,因此不在10X scRNA-seq/ST-seq的检测范围内。同样的,其他类型的小RNA,例如miRNA,也是10X scRNA-seq/ST-seq无法检测的。
13.10X scRNA-seq基础分析框架
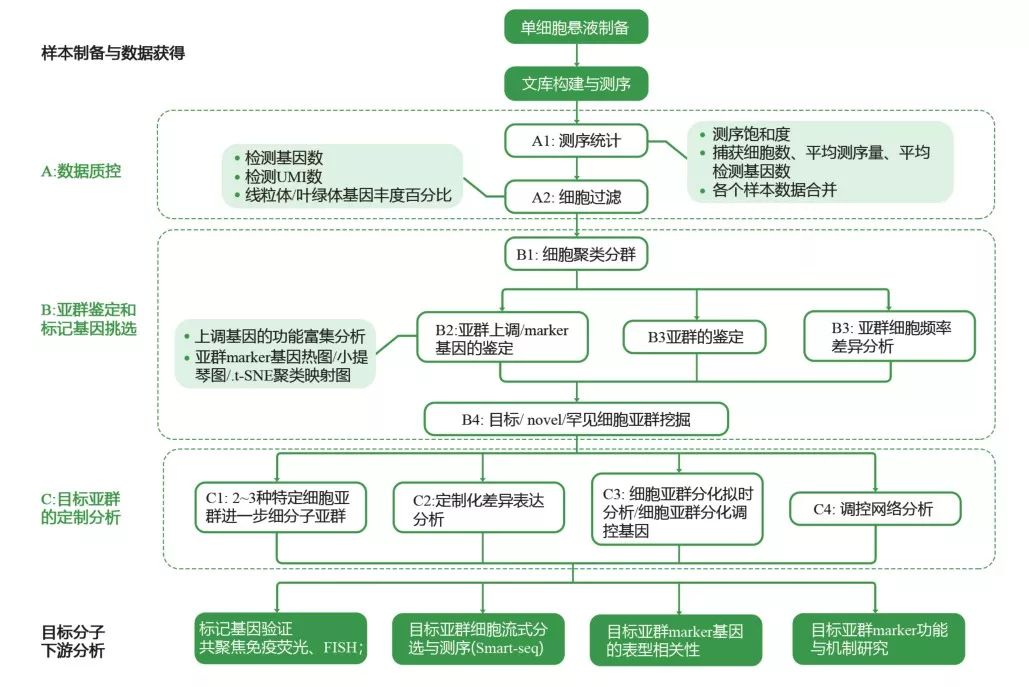
14.细胞过滤
(1)来源空载(油包水磁珠(GEM)数据):10X单细胞数据来源于一个油包水磁珠(GEM)为单元的单细胞扩增。约有65%的GEM在10X芯片的管道中可以成功捕获细胞,但也有35%没有捕获细胞。当然,空载的GEM并非没有任何结果。由于单细胞悬液中存在细胞破裂时产生的游离RNA,空载GEM中也会扩增得到少量的RNA信息,但基因数会很少。
(2)过载的GEM数据:部分GEM在10X芯片中可能也会出现过载,即一个GEM中吸入了两个或两个以上的细胞。这样两三个细胞信息就被合并了,从而相互干扰。
(3)状态异常的细胞信息:这部分常常是实验操作过程中衰亡自噬的细胞,这类细胞已经不具有研究的价值,应该被去除。
对于以上问题,主要通过以下三个指标进行过滤:
- (1)细胞中检测到的基因数,太低或者太高的细胞将被过滤;
- 检测基因数太少的单细胞数据,可能来自空载的GEM。检测基因数太高的,可能来自过载的GEM。
- (2)细胞中检测道德UMI总数,太低或者太高的细胞将被过滤;
- UMI就是cDNA扩增前给每个cDNA连上的一个标签序列。所以一段UMI序列,就代表一分子的cDNA。1个细胞UMI的总数就代表细胞中初始mRNA的总量。同样的,过滤mRNA总量太低或者太高的细胞,可以有效去除空载或过载的GEM。
- (3)线粒体基因表达量比例过高的细胞被去除
- 自噬凋亡的细胞,通常会出现线粒体基因的超表达。因此,利用这一指标,可以去除此类细胞。
15.细胞亚群分类的方法
分群的基本原理就是利用基因表达量的信息,计算各个细胞间表达模式的差异度,然后基于一定的标准将所有细胞归为多个亚群(将差异度小于特定值的细胞归为一个亚群)。分群是否符合预期,还需要后续基于特征标记对各个亚群进行定义后才可以判断。细胞分群结果一般要进行可视化展示(tSNE或UMAP图)。
16.亚群分类映射图
亚群分类结果展示,经常采用散点映射图的方式。这个过程可以分为两步:(1)先用某种算法(例如PCA或tSNE)将细胞间的关系用散点图进行展示;(2)根据某些指标(例如获得的细胞分群结果)将散点图中的点(代表细胞)涂上不同颜色,这个步骤称为映射。
17.tSNE算法与PCAE算法的比较
总的来说,PCA是常规转录组常用的数据降维和样本关系可视化的方法。但针对单细胞转录组数据,tSNE是明显胜过PCA的方法。
(1)PCA是线性降维的方法(适用于普通转录组)
以转录组数据为例,PCA的主要目的是从大量基因的表达量信息中,提取对整体基因表达量影响最大的效应(例如实验处理效应、实验批次效应、异常样本效应等)。这些效应被称为主成分(PC),然后利用主成分绘图。简而言之,只要2-3个变量(命名为PC1、PC2、PC3)就可以代表原来几万个基因含有的大部分信息。那么细胞之间表达量差异,就体现在PC1、PC2这些变量数值上的差异。最后,我们只要用PC1、PC2绘制散点图,就可以将有差异的细胞区分开。
PCA的方法侧重于去抓样本中隐含的主要效应,从而让差异大的样本在图中呈现较远的距离。在常规RNA-seq项目中,一般样本不多,实验处理效应组合数通常不会超过10种,因此每个实验处理效应在所有因素的总体效应中占比都比较大,属于效应比较大的因素。另外,实验批次效应、离群样本等也属于比较大的效应,这类效应易于被PCA获取。
但是如果影响样本分组的不是主要效应,而是一些效应更小的效应,PCA则无法对这些样本进行准确区分。而10X scRNA-seq的可视化展示,主要期望对各个细胞亚群有良好的区分。在每次检测的上万个细胞中,几乎肯定可以区分出几十种细胞亚群,包括一些稀有的细胞。区分这些细胞亚群(尤其是稀有细胞类别)的效应,往往不是主要效应(即大量基因的差异),而是一些微小效应(少量标记基因差异)。那么,PCA的方法就无法将这些细胞亚群良好区分。
(2)tSNE是非线性降维的方法(适用于单细胞转录组)
能够区分不同细胞亚群的理想算法,应兼顾以下两点:
- 局部结构:属于同一个亚群的细胞,聚类尽可能近; - 全局结构:属于不同亚群的细胞,聚类尽可能分开。
tSNE算法就属于上述这种可以同时兼顾局部结构和全局结构的非线性降维可视化算法。
tSNE不同于PCA(PCA主要目标是尽量去抓取群体中的主要效应),tSNE算法的主要目标是:在经历数据降维后(例如从2万个基因降低到2个变量),在原始2万个基因的数据集中最相似的细胞,在降维后的数据中依然保持最为相似(紧密成簇)。这就保证了哪怕稀有细胞只有少量基因区别于其他细胞亚群,在tSNE中依然可以与其他细胞有良好的区分。
18.其他类型的tSNE映射图
除了亚群分组的信息,细胞样本的来源信息也可以通过颜色的方式映射到tSNE图中,展示不同的生物学问题。另一种常见的映射方式就是以某个标记基因的表达量给各个点上色,从而可以直观观察该标记基因在哪些细胞亚群中表达。
19.细胞亚群的鉴定步骤
- (1)参考已有文献或数据库,确认样本所处的组织中常有的细胞类型有哪些,以及筛选对应细胞类型的已知标记基因(比如cell marker数据库);
- (2)将这些已知标记基因与我们自己测序数据中各个细胞亚群的上调/下调基因进行匹配,从而检测测序结果中各个细胞亚群所属的细胞类型(需要综合多个标记基因);
- (3)合并同类亚群:初步的亚群分类结果可能将同类细胞分成若亚类。基于分子标记,可以将归为同类细胞的亚群重新合并;
- (4)新标记基因筛选(其中包含的转录因子尤其值得关注);
- (5)新亚群的定义(潜在来源:低质量细胞、未被报道的新细胞、中间类型的过渡细胞)。
20.关于已知标记基因的筛选
可以使用哈尔滨医科大学整理的两个细胞标记基因数据库:
- (1)针对各个组织标记基因的cellMarker数据库;
- (2)针对肿瘤样本的CancerGEA数据库。