21.标记基因的展示
个位数标记基因可以选择小提琴图和tSNE映射图,十几个标记基因可以选择气泡图,大量标记基因可以选择热图。
22.亚群细胞频率
10X scRNA-seq通常会取多个样本,例如不同发育时期/不同病例发展时期的样本分别开展检测。在分析阶段,各个样本的数据将会被汇总后统一进行细胞分群。那么,各个样本中各类细胞相对比例的变化(某类细胞占总体细胞的百分比)也是非常重要的问题。
尤其是某些细胞亚群间,会存在上下游的演化分化关系。而如果知道细胞亚群之间潜在的演化关系,我们还可以用10X scRNA-seq数据模拟细胞的演化过程。这个分析就叫细胞分化轨迹分析或拟时间分析。
23.基于人工注释来鉴定细胞亚群
- (1)基于我们的预期(我们样本的类型)构建marker集,marker可以来自数据库或者文献;
- (2)已知marker基因与我们自己数据中的亚群特异表达基因进行匹配,初步鉴定每个亚群是什么类型的细胞;
- (3)使用逻辑推理+绘图可视化,去一一排除初步鉴定结果中矛盾的地方。
- 缺点:以上过程非常耗时间。
24.自动化细胞鉴定软件
目前已经又生物信息开发团队,开发出了一系列基于参考库(一般都是开发团队自己构建的参考库)进行自动化细胞鉴定的软件,包括celaref、SingleR(目前使用最多)、SuperCT、Garnett、cellassign、CHETAH等。这一类软件替代的是细胞注释过程的机械工作,也就是通过marker基因分布判断细胞亚群所属细胞类型。共有两类策略:一类是基于基因在不同细胞类型的表达量差异去判定marker基因;一类是基于人工提供的marker基因信息做后续分析。
类型一 基于基因表达量矩阵:
- (1)根据基因表达量矩阵寻找可靠的marker基因;
- (2)基于marker基因计算未鉴定细胞/细胞亚群与所有细胞类型的相关性
- (3)根据相关性进行细胞类型注释
SingleR以差异倍数来寻找marker基因,以spearman相关系数来计算相关性。celaref以差异基因来寻找marker基因,通过marker基因在待鉴定细胞亚群中上调倍数排名来体现相关性。CHETAH首先以细胞类型构建层级聚类树,将分支特异性基因作为marker基因,以细胞为某一条分支的可能性来体现相关性。
SingleR——目前使用最多的基于基因表达量矩阵的软件:节省计算资源,鉴定效果不俗。
类型二 基于既有marker基因:
这一类软件增加了人工在其中的占比,或者说增加了主观意识对细胞注释的影响,因为marker基因是由我们去主观定义的。这一类软件不再需要自行分析去寻找marker基因,而将主要的运算量用在了分类器的训练,后续使用分类器去进行细胞类型注释,其中使用最广泛的分类器训练方式就是人工神经网络模型,例如软件SuperCT和scANV1。
25.综合使用Seurat分群和singleR细胞鉴定结果
三点原则:
- (1)以Seurat分群为主,SingleR注释为辅;
- (2)少数服从多数;
- (3)如果疑义或者分歧,则继续细分。
依据解释如下:
- (1)原则一的依据:Seurat毕竟是直接利用测序数据在没有任何假设的情况下分群的结果,理论上可以避免任何认为主观判定导致的偏差。而SingleR的注释,还是依赖于我们选择哪些细胞作为参考库,较多受到认为主管选择的干扰。所以,Seurat的结果有更高的可信度。
- (2)原则二的依据:单细胞测序数据虽然总体比较准确,但数据量少,以及背景噪音的存在。所以少量细胞难以比米娜存在marker基因表达的波动,导致SingleR鉴定(使用较少的marker基因)结果中的假阳性。所以,在某一亚群SingleR鉴定结果中,各个类型细胞比例差异很大的情况下,遵从少数服从多数的原则。
- (3)原则三的依据:SingleR毕竟还是比较可靠的方法,如果SingleR得出的结果与Seurat出入很大,那么还是值得我们复查Seurat的结果去寻找潜在原因。
26.数据量与成本的矛盾
总的来说,细胞通量提高的同时,单个细胞的测序深度却在降低。
以10X单细胞转录组为例,每个细胞检测出的基因数普遍在1000-4000的水平。而实际上,每个细胞表达的基因数应该在1万-1.6万。那么从单个细胞水平看,很多基因是没有被检测到的。主要原因如下:(1)单个细胞的数据量相对比较少;(2)高通量的单细胞测序,无法保证100%捕获细胞中的所有转录本。
27.数据量对分群效果的影响
数据量对细胞分群几乎没有影响,单细胞测序的现有测序量完全可以满足细胞分群。10X单细胞分群一般是使用排名前10的主成分用于分群聚类,因此对每个细胞数据量并没有太高要求,每个细胞几千条reads就足够分群了。
28.数据量对差异分析的影响
在实际分析中,基因表达分析针对的目标是分群之后得到的细胞亚群。我们关注的目标不是某个细胞是否表达某个基因,是否表达量更高。而是关注某个亚群是否表达某个基因,某个基因在该亚群的表达量是否高于其他亚群。在这种情况下,我们实际上是把一个亚群作为关注的整体,而不是关注某个细胞。虽然在10X单细胞检测中,每个细胞数据里会随机丢失一些基因,但由于每个细胞的基因丢失是随机的,如果从整个细胞亚群来看,还是能够保证绝大部分基因被检测到(某些细胞里漏检的基因,在其他细胞里可以被检测到)。
关键结论:
- (1)分群分析的最小单位是细胞,但2-3k reads/细胞的数据量已经足以保证准确分群。
- (2)基因表达分析的最小单位实际上是细胞亚群。由于每个亚群一般至少有几十个细胞,20-50k reads/细胞的数据量,在某个50个细胞的亚群里,就相当于1000-2500k reads/亚群的数据量,已经大大超过达到基因检出饱和曲线平台期所需的数据量,足以保证亚群水平的基因表达分析的可靠性。
29.单细胞实验设计与分析
实验设计:
- (1)样本准备:选择什么组织,是否需要富集某种细胞,细胞悬液制备的难度;
- (2)数据分析:对预期采取的数据分析技术路线和方法有框架式的设计;
- (3)分子实验:预期下游要开展哪些分子实验。
可行性和创新性:
- (1)可行性:进行可行性论证,比如是否可以得到足够的组织量,细胞悬液制备的难度如何;
- (2)创新性:启动前做好文献调研和同业进展的调查,避免项目设计与已发表文献或正在进行的同行项目撞车。
30.单细胞实验中的样本准备
(1)个体状态 例如:不同疾病/疾病不同亚型;不同治疗方式;植物处于不同逆境胁迫条件。
(2)时间维度 例如:疾病/发育的不同时期。
(3)空间维度 例如:组织/亚组织部位(肿瘤组织不同位置)。
(4)细分细胞 深挖特点细胞类型,例如富集样本中的T细胞。
(5)组学结合 多组学贯穿分析,例如与单细胞VDJ-seq、单细胞ATAC-seq开展组学贯穿。
以上5个维度的组合,理论上可以组合出无数种研究方案,从而保证研究方案的独创性。
以肺部研究为例:
- (1)个体状态:可以选择正常肺部,肺纤维化,肺损伤,肺癌。
- (2)时间维度:例如研究肺部损伤,则可以取肺部损伤后的不同时间的样本。
- (3)空间维度:可以对细分的肺部不同部位取样,例如,支气管/肺泡;肺肿瘤组织不同位置取样。
- (4)细分细胞:肺全细胞图谱已有文章报道,但我们可以选择富集(例如,用荧光流式细胞技术筛选)某类细胞开展精细研究,例如间质细胞/上皮细胞;巨噬细胞;T细胞;I/II型肺泡细胞等,每个组织可以选择的细胞亚类很多。
- (5)组学组合:例如可以考虑10X scRNA-seq和10X ATAC-seq组合。
31.个性化分析
在获得10X单细胞转录组标准流程结果后,就可以考虑开展个性化分析了。
| 分析内容 | 分析依赖的生物学背景信息 |
|---|---|
| 特定亚群的进一步细分 | 前一阶段精确的细胞亚群鉴定 |
| 特定亚群(子亚群)的差异表达分析 | 1.前一阶段精确的细胞亚群/子亚群鉴定;2.比较组的设计 |
| 细胞分化轨迹/拟时间分析 | 1.前一阶段精确的细胞亚群鉴定;2.选择目标亚群;3.确定演化起点的细胞类型 |
| 基因调控网络分析 | 前一阶段精确的细胞亚群鉴定 |
32.目标亚群进一步细分
在初步完成细胞分类鉴定后,一般还会提取比较关注的2-3种细胞类型,进一步细分子亚群。
33.特定亚群(子亚群)的差异表达分析
在基础分析中,只是筛选每个类型细胞的标记基因(上调表达)。10X单细胞转录组本质还是转录组数据,只是样本数更高(每个细胞都作为一个样本),分组信息更加复杂。常见的转录组实验设计一般是两个变量因素,主要是个体维度(健康组vs患病组,品种A vs 品种B)和时间维度(不同发育阶段/病程发展不同阶段)。而10X单细胞转录组又加入了细胞亚群的维度,因此可以进行更丰富、精细的差异分析。
| 常规转录组 | 10X单细胞转录组 | |
|---|---|---|
| 比较目的 | 病人与正常人的差异 | 病人与正常人的差异 |
| 比较策略 | 病人、正常人组织间的差异分析 | 病人、正常人各个细胞亚群/子亚群间的差异分析 |
| 比较策略 | 反映整个组织转录组的平均差异,但仅在部分细胞亚群中存在的差异将可能被稀释掩盖 | 每个细胞亚群中存在的差异可以被挖掘,可能发现常规转录组遗漏的关键差异 |
34.细胞分化轨迹分析
- 分类式分析策略:细胞亚群分类分析、亚群基因表达差异分析。
- 系统化分析策略:亚群分化轨迹分析、基因表达调控网络分析。
基于转录组数据,利用模型预测细胞演化/分化过程,这样的研究策略一般称为细胞分化轨迹(differentiation trajectories)分析或轨迹推断(trajectories inference)。细胞分化轨迹分析的结果又常常被称为拟(伪)时间(Pseudotime)分析。
35.Monocel2细胞分化轨迹分析的主要步骤
STEP1:信息筛选
(1)细胞筛选:只筛选潜在存在分化关系的细胞亚群;
通常只有属于同一大类的细胞才会被纳入分析。
(2)基因筛选:只保留存在差异或变异较大的基因用于分析。
Monocel2提供了三种基因筛选策略:1)选择差异表达基因;2)选择高变异系数基因;3)选择预设的标记基因。
STEP2:轨迹构建
(1)降低维度;(2)构建细胞间分化的轨迹,并将每个细胞映射到路径中。
STEP3:寻找细胞分化相关的基因
36.新细胞类型解析的步骤
(1)识别和鉴定(依赖标记基因):
1)已知细胞的子亚型:对应子亚型下的标记基因特性。
2)过渡细胞:基于分化轨迹分析。
3)全新亚型:需要参考更广泛的数据,甚至其他组织、近缘物种的细胞数据。
(2)推测功能
基于新亚群标记基因的功能,可以初步推断对应亚群的功能。一般需要关注相关标记基因在哪个疾病/形状研究中被报道过,以及可以多关注转录因子。
(3)功能验证:细胞实验、动物实验
37.Seurat相关参数的解读
来源:冻春卷的简书
pbmc <- CreateSeuratObject(counts=pbmc.data,project="pbmc2700",min.cells = 3,min.features = 200)
在使用CreateSeuratObject()创建对象的同时,过滤数据质量差的细胞。保留在>=3个细胞中表达的基因;保留能够检测到>=200个基因的细胞。
counts:raw counts or TPMs;project:设置项目名称。
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
查看上述三个基因在在前30个细胞中的数据
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
添加线粒体百分比列。用 [[]] 可以在对象的metadata中添加列,中间则是列名
pbmc <- ScaleData(pbmc, features = all.genes)
缩放数据:给予下游分析同等的权重,这样高表达基因就不会占主导地位。
- (1)使每个基因在所有细胞间的表达量均值为0;
- (2)使每个基因在所有细胞间的表达量方差为1。
缩放是Seurat工作流程中的一个重要步骤,但仅限于将要进行PCA的基因。因此,ScaleData中的默认值只是对前面确定的变量特性(默认值为2,000)执行缩放。要做到这一点,可以忽略前面函数调用中的features参数,即pbmc <- ScaleData(pbmc)。
由于Seurat heatmap(与DoHeatmap一起产生,如下图所示),需要对heatmap中所有基因进行缩放,以确保高表达的基因不会在heatmap中占主导地位。为了确保在后面的热图中不会遗漏任何基因,所以也可缩放所有基因。
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
使用ScaleData函数从单细胞数据集中删除不需要的变量。例如,我们可以“regress out”与细胞周期阶段或线粒体污染相关的异质性(去除不需要的变量,即校正协变量,校正线粒体基因比例的影响)。
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
对上一步骤的变量得到的缩放数据进行线性降维分析(PCA)。
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
DimHeatmap可以探索数据集的异质性来源,帮助确定用于下游分析的主成分。细胞和features都是根据它们的PCA分数排序。dims=1:15是指展示前15个PC。

可以看到PC1-6对于数据分离比较有帮助,而越往后面帮助越小,可以通过这种方式来确定,下游分析中用于分析的主成分。
pbmc <- JackStraw(pbmc, num.replicate = 100) # 重复一百次
pbmc <- ScoreJackStraw(pbmc, dims = 1:20) # 选择20个PC
为了克服scRNA-seq数据的technical noise,Seurat基于它们的PCA分数对单元进行分组,每个PC实质上代表一个“metafeature”,它将跨相关特征集的信息组合在一起。后续,使用JackStraw算法来确定数据集的维度,即该选多少个PC才合适。
上述代码解读:1)随机置换一部分数据(默认为1%),然后重新 PCA,重复100。将包含较多低 P 值特征的主成分为“显著的”主成分;2)选择20个PC;3)JackStrawPlot函数提供了一个可视化工具,用于用均匀分布(虚线)比较每个PC的p值分布。
JackStrawPlot(pbmc, dims = 1:15)
“显著的”PCs将显示出一个低p值(虚线以上的实线)的强富集特性。在这种情况下,在最初的10-12个pc之后,重要性似乎急剧下降。
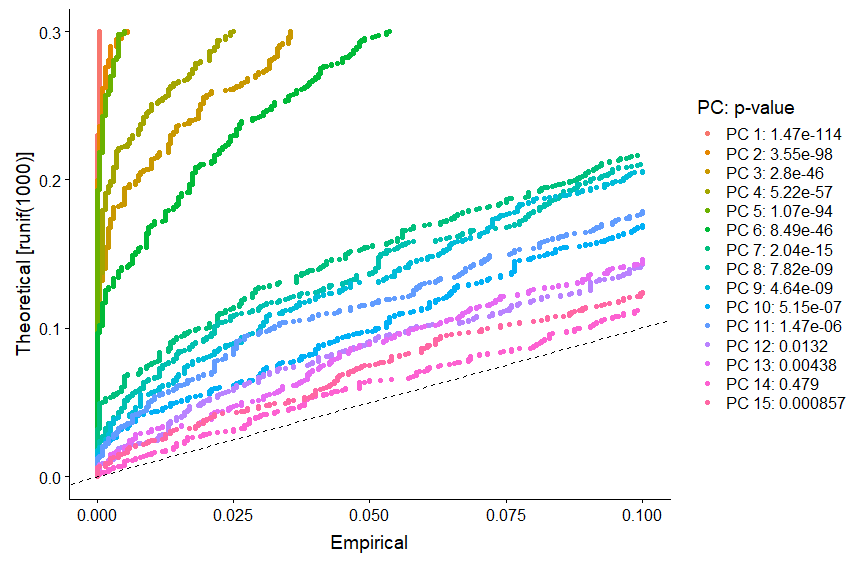
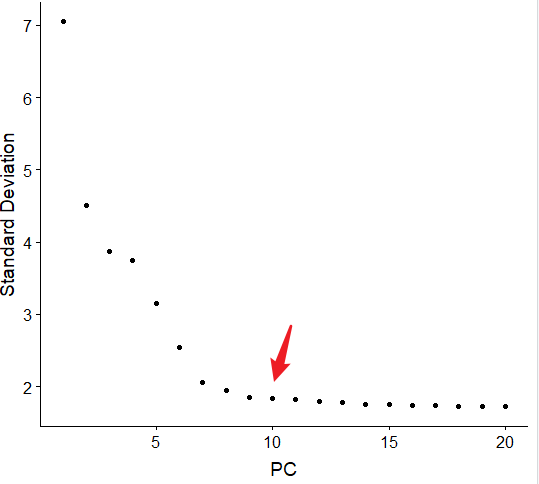
ElbowPlot(pbmc)
根据每个组成部分的方差百分比对主要组成部分的方差结实率进行排序(弯头图函数)。在这个例子中,我们可以观察到PC9-10周围的一个“弯头”,这表明大部分真实信号是在前10个pc中捕获的。
pbmc <- FindNeighbors(pbmc, dims = 1:10) # 前10个PC
pbmc <- FindClusters(pbmc, resolution = 0.5) # 分辨率是0.5
Cluster the cells 细胞聚类:
- (1)先在PCA空间中构造一个基于欧氏距离的KNN图,然后根据任意两个细胞在局部邻近区域的共享重叠(Jaccard相似性)来细化它们之间的边权值。此步骤使用FindNeighbors函数执行,并将之前定义的数据集维度(前10个pc)作为输入。
- (2)为了对细胞进行聚类,应用模块化优化技术,如Louvain算法(默认)或SLM,以迭代方式将单元分组在一起,目标是优化标准模块化函数。FindClusters函数实现这个过程,并包含一个分辨率参数,该参数设置下游集群的“granularity”,增加的值将导致更多的集群。将该参数设置在0.4-1.2之间,对于3K左右的单细胞数据集通常会得到良好的结果。对于较大的数据集,最佳分辨率通常会增加。可以使用Idents函数找到clusters。
# 安装UMAP
reticulate::py_install(packages ="umap-learn")
pbmc <- RunUMAP(pbmc, dims = 1:10) # 102.7 Mb
DimPlot(pbmc, reduction = "umap", pt.size=1)
# 可视化,用umap的方法
# 保存数据
saveRDS(pbmc, file = "../output/pbmc_tutorial.rds")
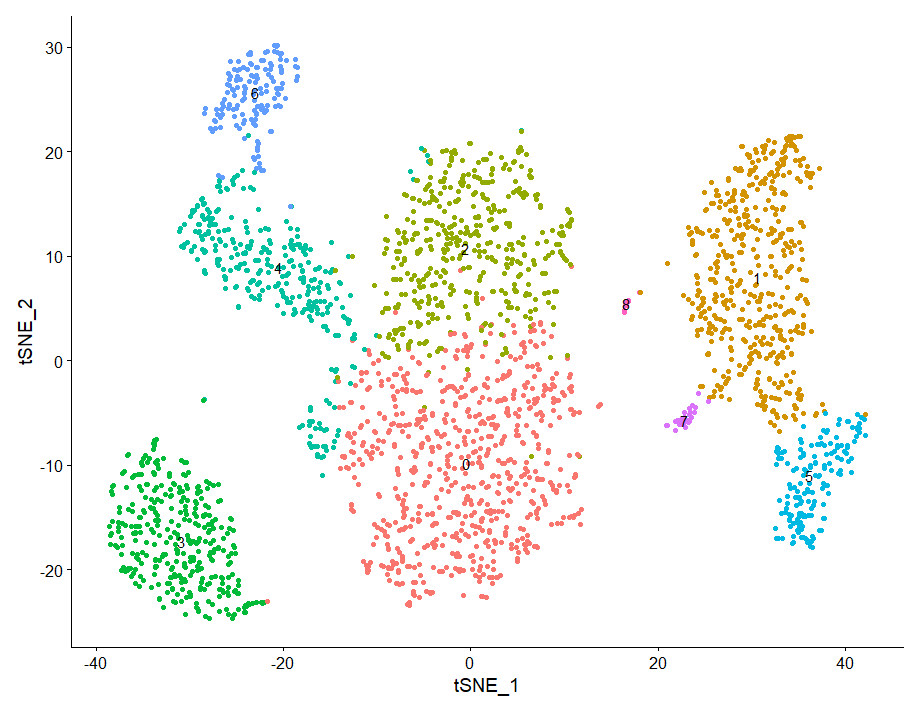
Seurat提供了几种非线性的降维技术,如tSNE和UMAP,以可视化和探索这些数据集。对于UMAP和tSNE的输入数据,我们建议使用相同的PCs作为聚类分析的输入。
# 找cluster 1的markers
cluster1.markers <- FindMarkers(pbmc,
ident.1 = 1, # 至少检测到1个feature
min.pct = 0.25) # 设置后会加速,默认是0.1
# 耗时23 s
head(cluster1.markers, n = 5) # 显示!
寻找亚群的marker基因:Seurat可以找到通过差异表达式定义集群的标记。默认情况下,它识别单个簇的阳性和阴性标记(在ident1中指定),与所有其他细胞相比较。findallmarker为所有集群自动化这个过程,但是您也可以测试集群组之间的相互关系,或者测试所有细胞。
- (1) min.pct参数要求在两组单元中至少检测到一个特征;
- (2) thresh.test参数要求一个特性在两组之间有一定的差异(平均);若两个值都设置为0,会非常耗时——因为这将测试大量不太可能具有高度差异性的features;
- (3) 为了加速,可以设置max.cells.per.ident参数,这将向下采样每个标识类,使其单元格数不超过设置的单元格数。虽然通常会有功率的损失,速度的增长可能是显著的,最高度差异表达的特征可能仍然会上升到顶部。
37.单细胞常用的数据结构SingleCellExperiment以及scRNA-seq workflow
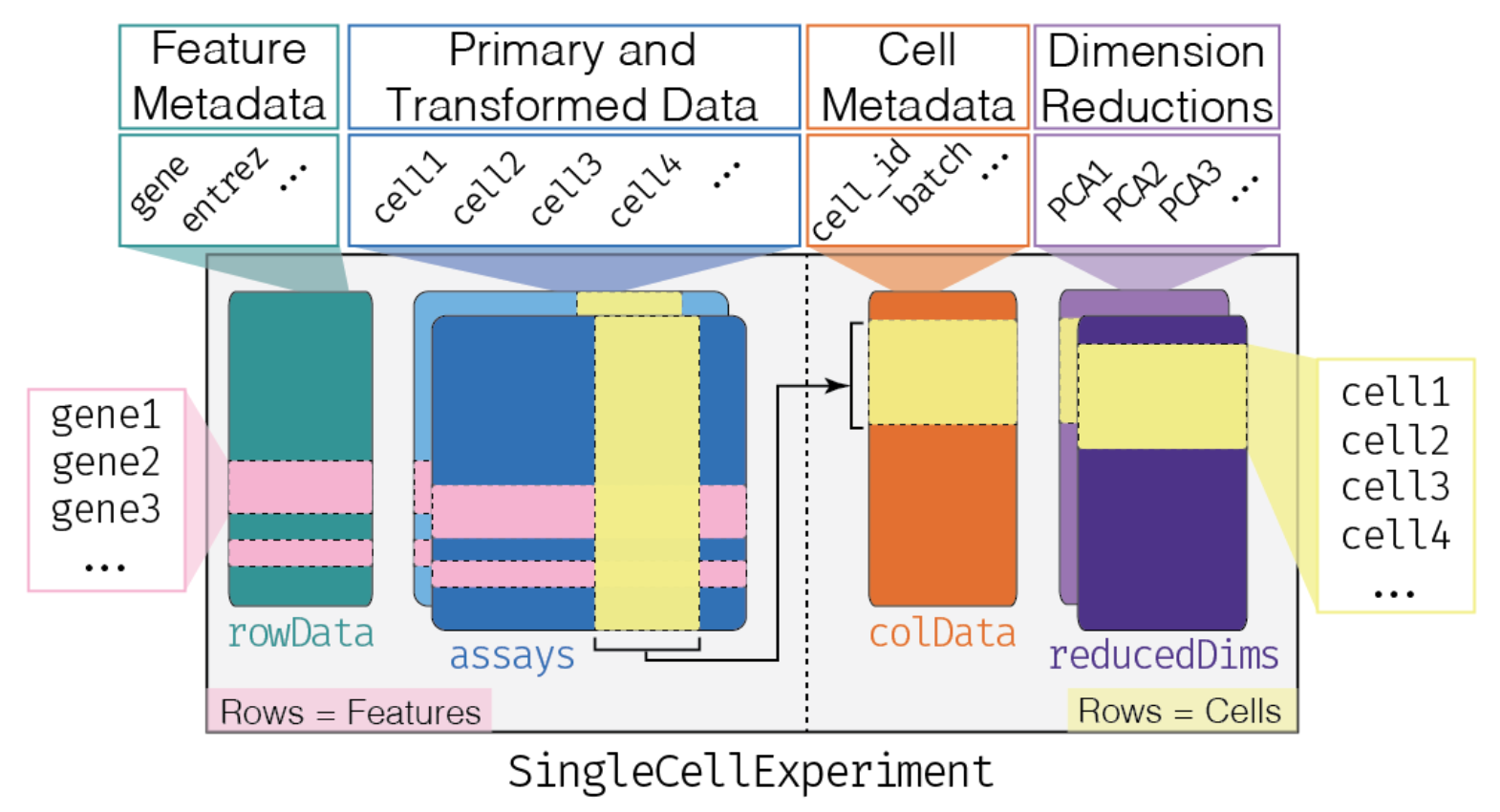
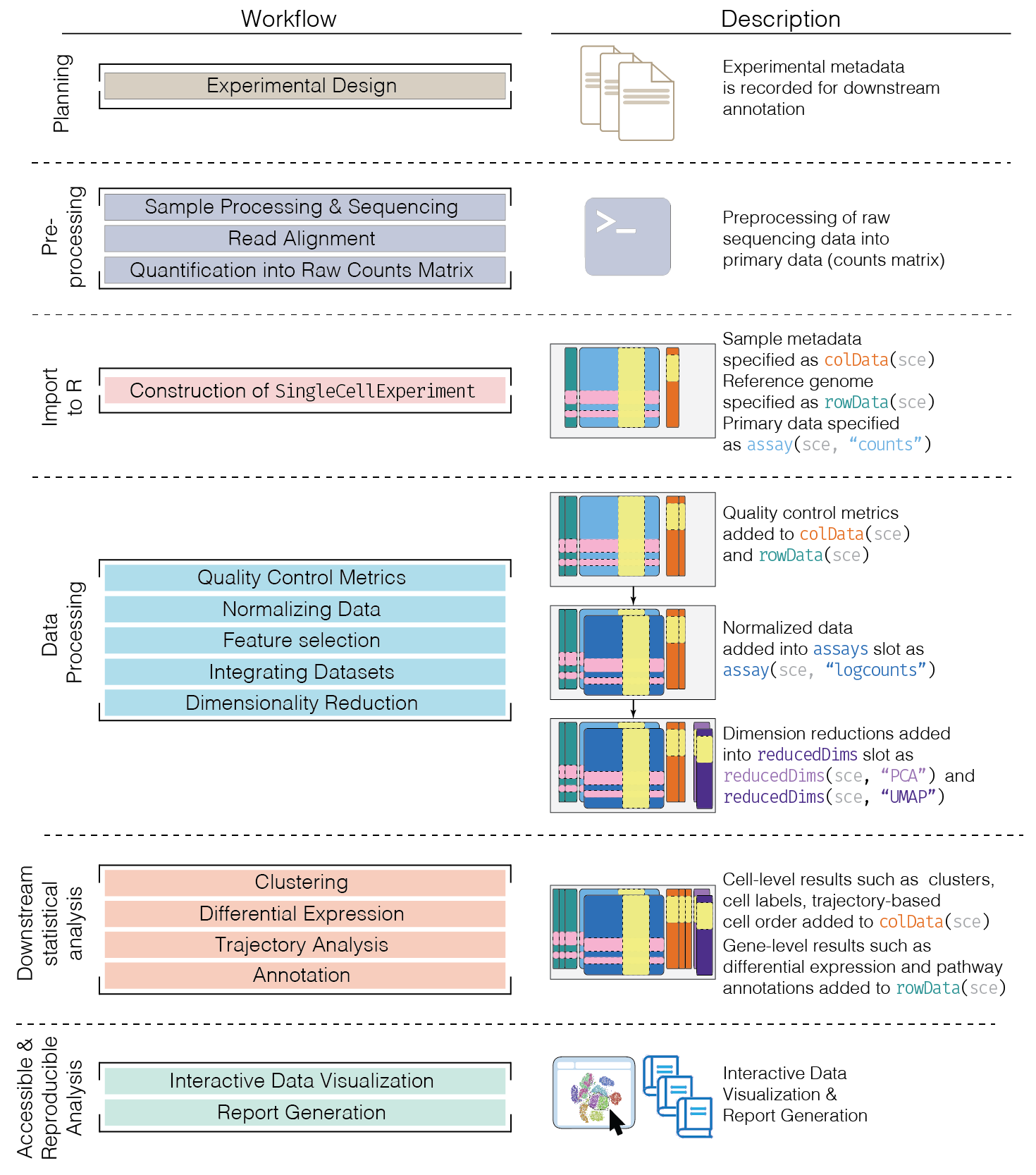
划重点:数据处理与下游分析
- 首先进行质控:去掉低质量细胞。这些细胞可能在建库环节被破坏,可能没有被有效捕获(这就是所谓的“dropout”)。一般会统计:每个细胞的全部count数、spike-in或线粒体reads比例、检测到基因的数量
- 表达矩阵归一化:为了减小细胞文库的偏差(可能由于细胞捕获效率不同、测序深度的差异而造成文库大小差异),把细胞们放在同一起跑线上,才能进行下面的细胞相似性比较,后面再根据相似性进行细胞分群。一般是基于log转换(当然有的函数也涉及了一些size factor的计算),从而对均值-方差进行校正
- 挑选一些特征基因(一般是高变化基因HVGs,Highly Variable Genes)进行下游分析。原理是根据每个基因在细胞之间的差异构建变化模型,然后找那些变化差异大的基因。使用HVGs不用全部基因的原因一是为了减少计算量,二是减少不感兴趣基因(比如在细胞之间没什么差异)对分析产生的噪音
- 降维处理:让数据更“紧凑”,一般是线性降维PCA+非线性降维tSNE/umap。PCA一般是先获得初步的低维数据(可能会挑出几十个主成分),然后传给t-SNE进一步压缩,进行可视化
- 细胞聚类:根据细胞归一化后的表达量相似性分成组,然后根据每个组marker基因(可理解为这一群细胞的标志性基因)的差异表达对分群进行生物学定义
38.单细胞数据处理——Normalization与Scaling
-
Normalization “normalizes” within the cell for the difference in sequenicng depth / mRNA throughput. 主要着眼于样本的文库大小差异 。(归一化,归的是技术误差,而不是批次效应)
-
Scaling “normalizes” across the sample for differences in range of variation of expression of genes . 主要着眼于基因的表达分布差异。
-
一般来说,应该先进行normalization,再进行scale,而scale的结果会用来后续的降维和聚类。
-
在Seurat中,一般得到原始表达矩阵并过滤后,会进行NormalizeData(),当然也可以使用自己的归一化结果(例如TPM结果,只是需要再log一下);在挑选HVGs之后,降维处理之前,还需要用到ScaleData()进行标准化。对每个进行center后的值再除以标准差(就是进行了一个z-score的操作)。
39.根据基因取细胞子集

也可以提取特定Cluster,进行进一步细分:
# 提取特定cluster,继续后续分析。
ident_df <- data.frame(cell=names(Idents(pbmc)), cluster=Idents(pbmc))
pbmc_subcluster <- subset(pbmc, cells=as.vector(ident_df[ident_df$cluster=="Naive CD4 T",1]))
40.RunTSNE不是在聚类
-
聚类(clustering)是直接基于距离矩阵的经典无监督机器学习问题。通过最小化簇内距离或在降维后的表达空间中鉴定密集区域,将细胞分配给簇。方法有层级聚类、K-menas、基于图的聚类等 (基因共表达聚类分析及可视化,基因表达聚类分析之初探SOM)。
-
tSNE和UMAP目前主要用于可视化。用于可视化的降维必然涉及信息丢失并改变细胞之间的距离。因此tSNE/UMAP图应仅只用于解释或传达基于更精确的、更多维度的定量分析结果。这样可以保证分析充分利用了压缩到二维空间时丢失的信息。假如二维图上呈现的细胞分布与使用更多数目的PC进行聚类获得的结果之间存在差异,应倾向于相信后者(聚类)的结果