资料来源:公众号:育种数据分析之放飞自我
1.前言
这里,总结一下GWAS的学习笔记,GWAS全称“全基因组关联分析”,使用统计模型找到与性状关联的位点,用于分子标记选择(MAS)或者基因定位,这次学习的教程是plink做GWAS,plink是个很好的软件,但是我之前做GWAS都是使用R包,听说plink和EMMAX做GWAS更快,更好,更容易写出pipeline。就利用网上的信息写一个操作笔记,先操作plink,然后是EMMAX。对于一些有模型基础的同学,理解起来应该不难。
1.1 GWAS分析的两类性状:
- 分类性状(阈值性状,质量性状):比如抗病性,颜色等等
- 连续性状(数量性状):比如株高,体重,产量等等
1.2 GWAS的分析方法:
- 分类性状:logistic等等
- 连续性状:GLM,MLM模型等等
(1)一般线性模型(GLM):
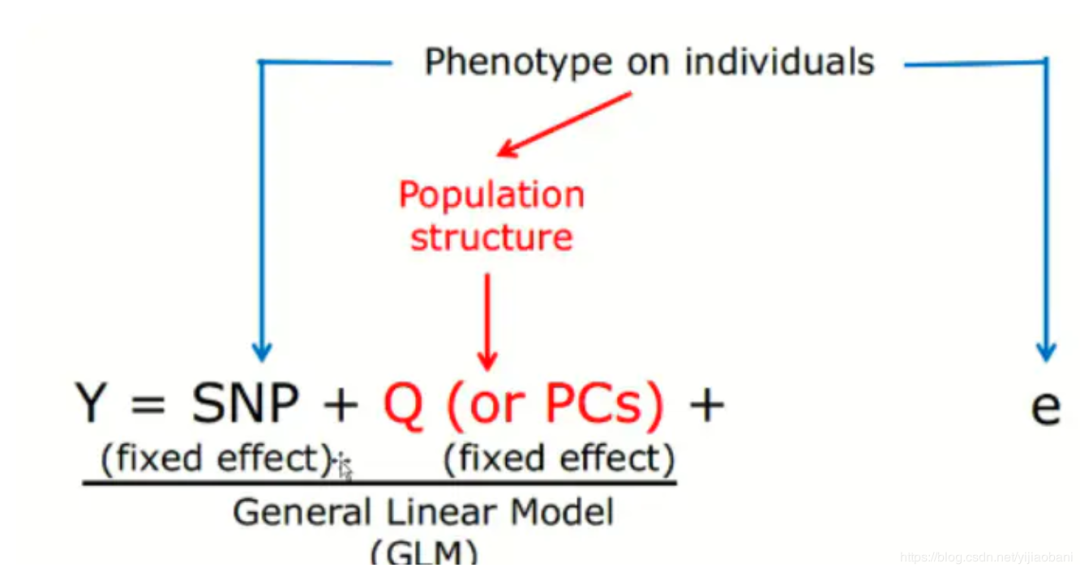
这里,SNP作为固定因子,可以考虑其它协变量(比如性别,PCA,群体结构等等)
(2)混合线性模型(MLM):
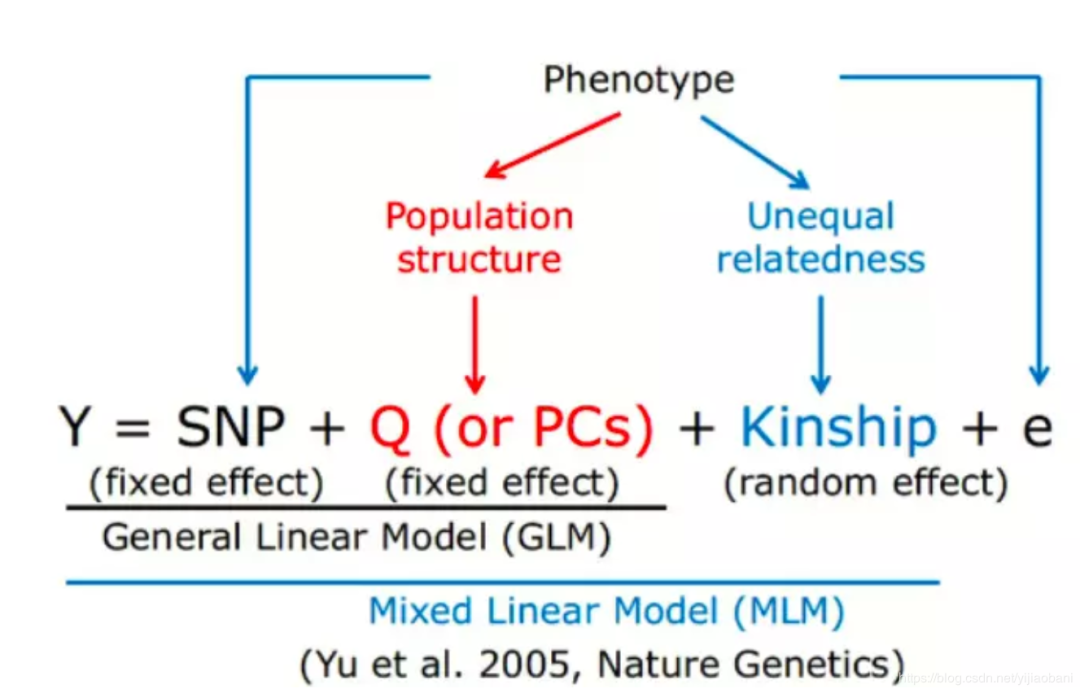
固定因子:SNP + 可以考虑其它协变量(比如性别,PCA,群体结构等等),这里固定因子和前面的GLM一样 随机因子:亲缘关系矩阵(K矩阵或者A矩阵)
2.下载数据和代码
首先,在linux环境下,新建一个文件夹,进入后运行下面命令:
git clone https://github.com/MareesAT/GWA_tutorial.git
下载之后,目录如下:
3.下载R语言和plink软件
- R:https://www.r-project.org/
- plink:http://zzz.bwh.harvard.edu/plink/ https://www.cog-genomics.org/plink2
4.解压文件
unzip 1_QC_GWAS.zip
unzip 2_Population_stratification.zip
unzip 3_Association_GWAS.zip
5.文件介绍
5.1 质控
主要是根据一些筛选标准,去掉一些位点。筛选标准有缺失百分比,哈温等等。
文件夹:1_QC_GWAS
主要文件:

其中,1_Main_script_QC_GWAS.txt里面包括所有运行的代码,.HapMap文件是plink格式的文件,.R是几个用于检测和可视化的R脚本,我们后面会依次讲解这些代码。
5.2 群体分层
文件夹:2_Population_stratification

其中,1_Main_script_QC_GWAS.txt,2_Main_script_MDS.txt为运行代码。
5.3 GWAS分析
文件夹:3_Association_GWAS

其中,其中,3_Main_script_association_GWAS.txt为运行代码。
5.4 多基因风险评分(PRS)分析
是一个word文件:
这是独立的一步,有需要的可以查看里面的分析方法。
