资料来源:公众号:育种数据分析之放飞自我
1.前言
上一次我们经过去掉缺失,去掉错误的性别信息,得到的文件为:
这里,我们根据最小等位基因频率(MAF)去筛选。
为什么要根据MAF去筛选?
最小等位基因频率怎么计算?比如一个位点有AA或者AT或者TT,那么就可以计算A的基因频率和T的基因频率,qA + qT = 1,这里谁比较小,谁就是最小等位基因频率,比如qA = 0.3, qT = 0.7, 那么这个位点的MAF为0.3. 之所以用这个过滤标准,是因为MAF如果非常小,比如低于0.02,那么意味着大部分位点都是相同的基因型,这些位点贡献的信息非常少,增加假阳性。更有甚者MAF为0,那就是所有位点只有一种基因型,这些位点没有贡献信息,放在计算中增加计算量,没有意义,所以要根据MAF进行过滤。
2.去掉性染色体上的位点
思路:
- 在map文件中选择常染色体,提取snp信息
- 根据snp信息进行提取
提取常染色体上的位点名称:因为这里是人的数据,所以染色体只需要去1~22的常染色体,提取它的家系ID和个体ID,后面用于提取。
awk '{ if($1 >=1 && $1 <= 22) print $2}' HapMap_3_r3_6.bim > snp_1_22.txt
wc -l snp_1_22.txt
1399306 snp_1_22.txt
常染色体上共有1399306位点。
3.提取常染色体上的位点
这里,用到了位点提取参数–extract:
plink --bfile HapMap_3_r3_6 --extract snp_1_22.txt --make-bed --out HapMap_3_r3_7
查看日志:
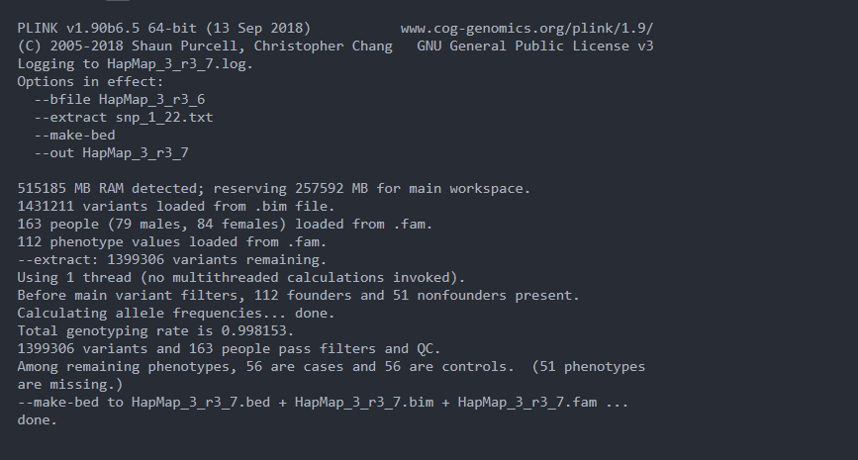
可以看到,共有163个基因型,共有1399306个SNP。
4.计算每个SNP位点的基因频率
首先,通过参数–freq,计算每个SNP的MAF频率,通过直方图查看整体分布。可视化会更加直接。
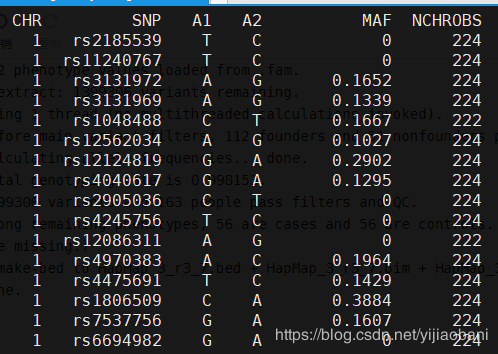
plink --bfile HapMap_3_r3_7 --freq --out MAF_check
结果文件:MAF_check.frq预览:
5.对基因频率作图
maf_freq <- read.table("MAF_check.frq", header =TRUE, as.is=T)
pdf("MAF_distribution.pdf")
hist(maf_freq[,5],main = "MAF distribution", xlab = "MAF")
dev.off()
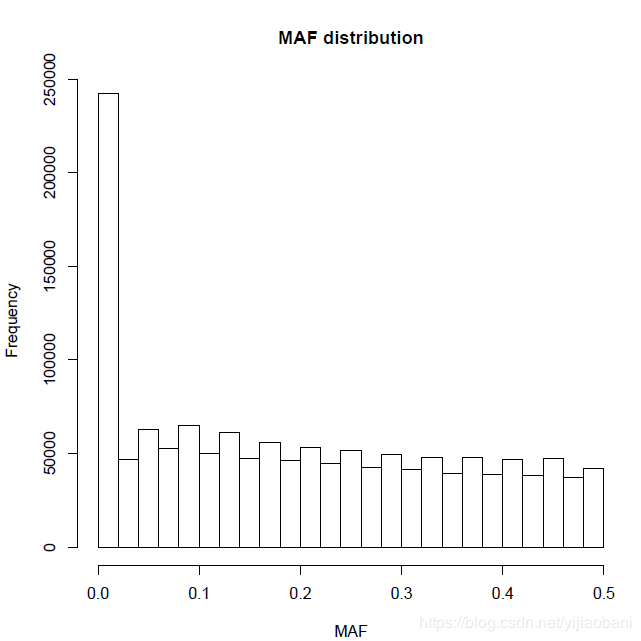
可以看出,很多基因频率为0,说明没有分型,这些位点需要删掉。
6.去掉MAF小于0.05的位点
plink --bfile HapMap_3_r3_7 --maf 0.05 --make-bed --out HapMap_3_r3_8
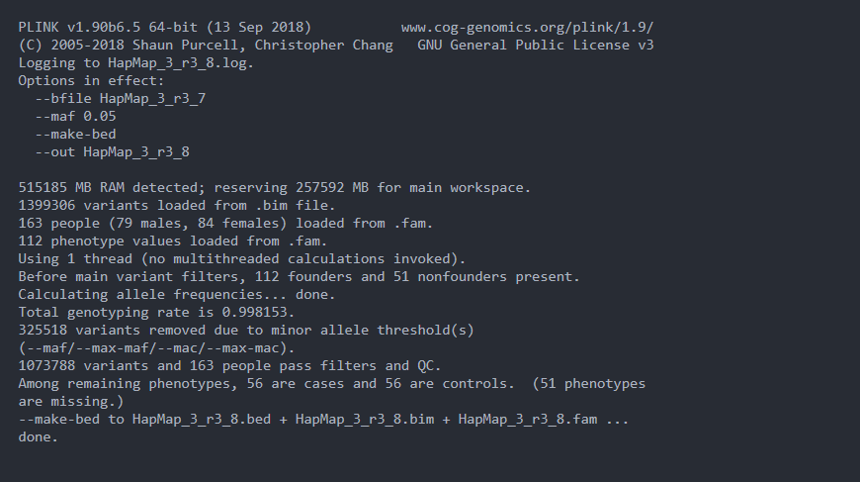
可以看到,325518个位点被删掉了,剩余1073788 个位点。
结果文件如下,后面我们用这个文件,进行后续的质控。
