资料来源:公众号:育种数据分析之放飞自我
1.前言
一般自然群体,基因型个体的杂合度过高或者过低,都不正常,我们需要根据杂合度进行过滤。偏差可能表明样品受到污染,近亲繁殖。我们建议删除样品杂合率平均值中偏离±3 SD的个体。
参数过滤和手动过滤:plink有个特点,所有的过滤标准,都可以生成过滤前的文件,然后可以手动过滤,也可以用参数进行过滤。
- 比如:–missing生成结果,也可以用–geno和–mind过滤。
- 比如:–hardy生成结果,可以使用–hwe过滤
- 比如:–freq生成结果,可以用–maf过滤 但是杂合度–het,没有过滤的函数,只能通过编程去提取ID,然后用–remove去实现。
2.计算杂合度
plink --bfile HapMap_3_r3_9 --het --out R_check
结果文件:
.het (method-of-moments F coefficient estimates)
Produced by --het.
A text file with a header line, and one line per sample with the following six fields:
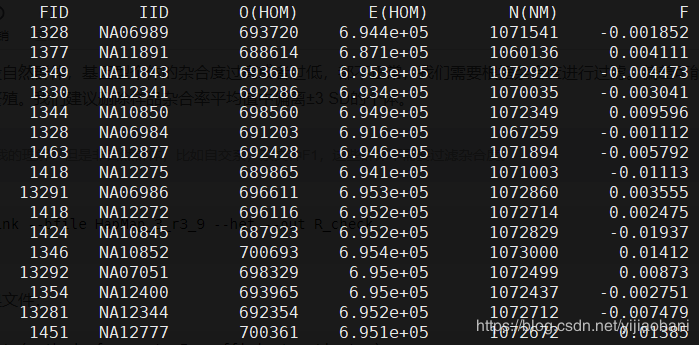
FID Family ID # 家系ID
IID Within-family ID # 个体ID
O(HOM) Observed number of homozygotes # 实际纯合个数
E(HOM) Expected number of homozygotes # 期望纯合个数
N(NM) Number of non-missing autosomal genotypes # 总个数
F Method-of-moments F coefficient estimate #
所以,杂合度 = (N-O)/N
3.杂合度可视化
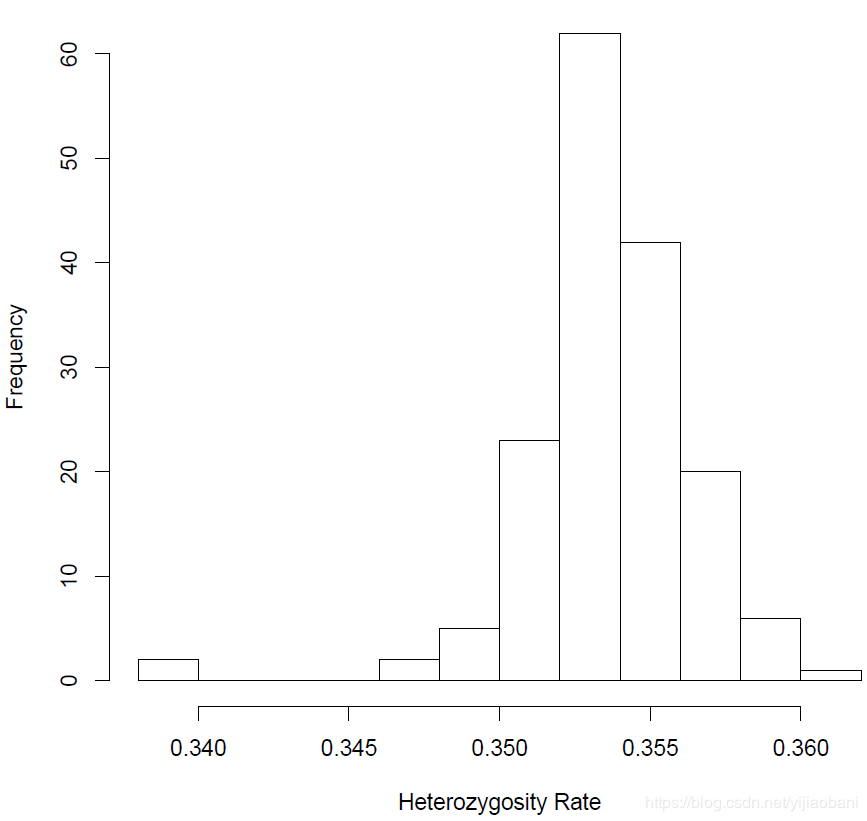
het <- read.table("R_check.het", head=TRUE)
pdf("heterozygosity.pdf")
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
hist(het$HET_RATE, xlab="Heterozygosity Rate", ylab="Frequency", main= "Heterozygosity Rate")
dev.off()
4.计算杂合度三倍标准差以外的个体
首先,查看哪些个体在3倍标准差以外:
het <- read.table("R_check.het", head=TRUE)
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
het_fail = subset(het, (het$HET_RATE < mean(het$HET_RATE)-3*sd(het$HET_RATE)) | (het$HET_RATE > mean(het$HET_RATE)+3*sd(het$HET_RATE)));
het_fail$HET_DST = (het_fail$HET_RATE-mean(het$HET_RATE))/sd(het$HET_RATE);
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE)
结果可以看出,这两个个体杂合度在3倍标准差以外:

5.去掉这些个体
cat fail-het-qc.txt
"FID" "IID" "O.HOM." "E.HOM." "N.NM." "F" "HET_RATE" "HET_DST"
1330 "NA12342" 703770 691000 1066256 0.03402 0.33996151018142 -4.75272486494316
1459 "NA12874" 709454 695300 1072858 0.03758 0.338725162137021 -5.18288854902555
先对数据进行清洗,去掉引号,然后提取家系和个体ID
sed 's/"//g' fail-het-qc.txt |awk '{print $2}' > het_fail_ind.txt
sed 's/"//g' fail-het-qc.txt |awk '{print $1,$2}' > het_fail_ind.txt
使用remove去掉这两个个体
plink --bfile HapMap_3_r3_9 --remove het_fail_ind.txt --make-bed --out HapMap_3_r3_10
6.结果文件
