资料来源:公众号:育种数据分析之放飞自我
1.前言
这里,我们要对一些亲子关系的个体,进行一下过滤,计算类似IBS的结果。这里讲亲子关系的个体移除,不是必须要的,比如我们分析的群体里面有亲子关系的个体,想要进行分析,不需要做这一步的筛选。
2.计算pihat > 0.2的组合
plink --bfile HapMap_3_r3_10 --genome --min 0.2 --out pihat_min0.2
说明文档:
--genome invokes an IBS/IBD computation, and then writes a report with the following fields to plink.genome:
FID1 Family ID for first sample
IID1 Individual ID for first sample
FID2 Family ID for second sample
IID2 Individual ID for second sample
RT Relationship type inferred from .fam/.ped file
EZ IBD sharing expected value, based on just .fam/.ped relationship
Z0 P(IBD=0)
Z1 P(IBD=1)
Z2 P(IBD=2)
PI_HAT Proportion IBD, i.e. P(IBD=2) + 0.5*P(IBD=1)
PHE Pairwise phenotypic code (1, 0, -1 = AA, AU, and UU pairs, respectively)
DST IBS distance, i.e. (IBS2 + 0.5*IBS1) / (IBS0 + IBS1 + IBS2)
PPC IBS binomial test
RATIO HETHET : IBS0 SNP ratio (expected value 2)
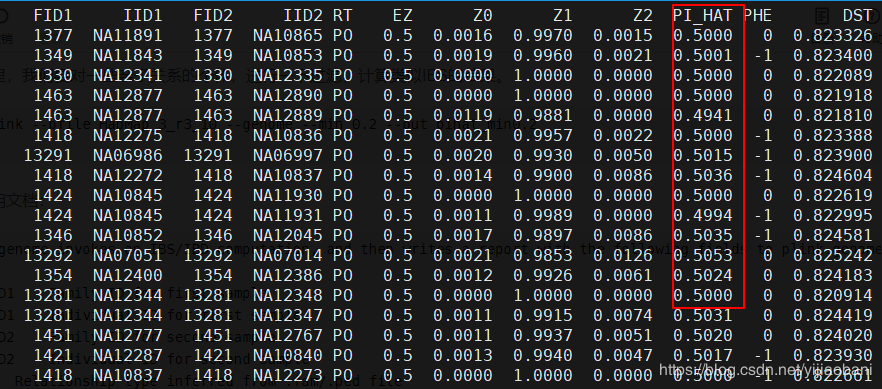
3.提取Z1大于0.9的个体
awk '{if($8>0.9) print $0}' pihat_min0.2.genome > zoom_pihat.genome
过滤出91个组合:
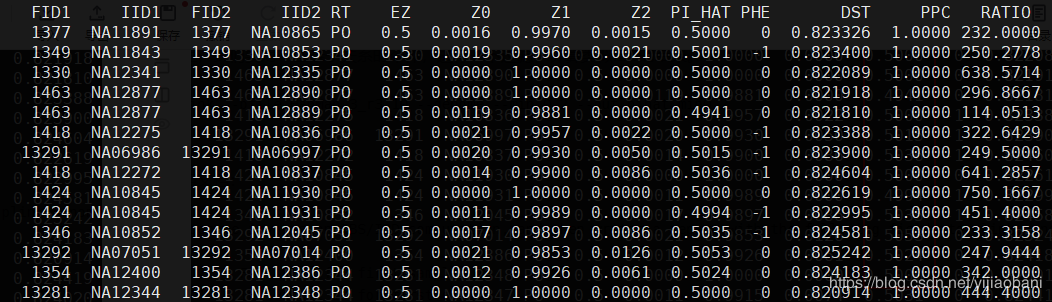
4.作图
pdf("relatedness.pdf")
relatedness = read.table("pihat_min0.2.genome", header=T)
par(pch=16, cex=1)
with(relatedness,plot(Z0,Z1, xlim=c(0,1), ylim=c(0,1), type="n"))
with(subset(relatedness,RT=="PO") , points(Z0,Z1,col=4))
with(subset(relatedness,RT=="UN") , points(Z0,Z1,col=3))
legend(1,1, xjust=1, yjust=1, legend=levels(relatedness$RT), pch=16, col=c(4,3))
pdf("zoom_relatedness.pdf")
relatedness_zoom = read.table("zoom_pihat.genome", header=T)
par(pch=16, cex=1)
with(relatedness_zoom,plot(Z0,Z1, xlim=c(0,0.02), ylim=c(0.98,1), type="n"))
with(subset(relatedness_zoom,RT=="PO") , points(Z0,Z1,col=4))
with(subset(relatedness_zoom,RT=="UN") , points(Z0,Z1,col=3))
legend(0.02,1, xjust=1, yjust=1, legend=levels(relatedness$RT), pch=16, col=c(4,3))
pdf("hist_relatedness.pdf")
relatedness = read.table("pihat_min0.2.genome", header=T)
hist(relatedness[,10],main="Histogram relatedness", xlab= "Pihat")
dev.off()
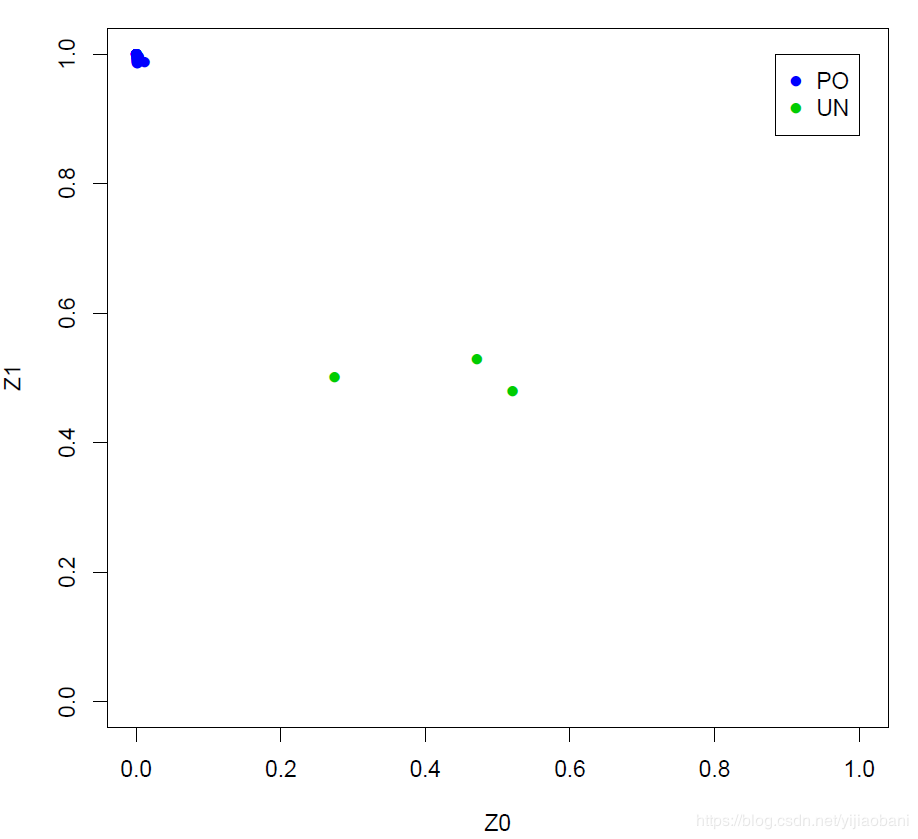
这里的PO为:亲子关系 这里的UN为:非亲缘关系
5.删除亲子关系的个体
plink --bfile HapMap_3_r3_10 --filter-founders --make-bed --out HapMap_3_r3_11
日志:
PLINK v1.90b6.5 64-bit (13 Sep 2018) www.cog-genomics.org/plink/1.9/
(C) 2005-2018 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to HapMap_3_r3_11.log.
Options in effect:
--bfile HapMap_3_r3_10
--filter-founders
--make-bed
--out HapMap_3_r3_11
515185 MB RAM detected; reserving 257592 MB for main workspace.
1073743 variants loaded from .bim file.
161 people (77 males, 84 females) loaded from .fam.
110 phenotype values loaded from .fam.
51 people removed due to founder status (--filter-founders).
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 110 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate in remaining samples is 0.998016.
1073743 variants and 110 people pass filters and QC.
Among remaining phenotypes, 55 are cases and 55 are controls.
--make-bed to HapMap_3_r3_11.bed + HapMap_3_r3_11.bim + HapMap_3_r3_11.fam ...
done.
可以看出,51个个体被移除。
6.结果文件

7.注意
这里讲亲子关系的个体移除,不是必须要的,比如我们分析的群体里面有亲子关系的个体,想要进行分析,不需要做这一步的筛选。