资料来源:公众号:育种数据分析之放飞自我
本篇主要是介绍一般线性模型(LM)中的linear参数,考虑因子协变量,然后将结果与R语言编程结果比较。
1.协变量文件整理
第一列为FID, 第二列为ID ,第三列以后为协变量(注意,只能是数值,不能是字符!)
这里协变量文件为:
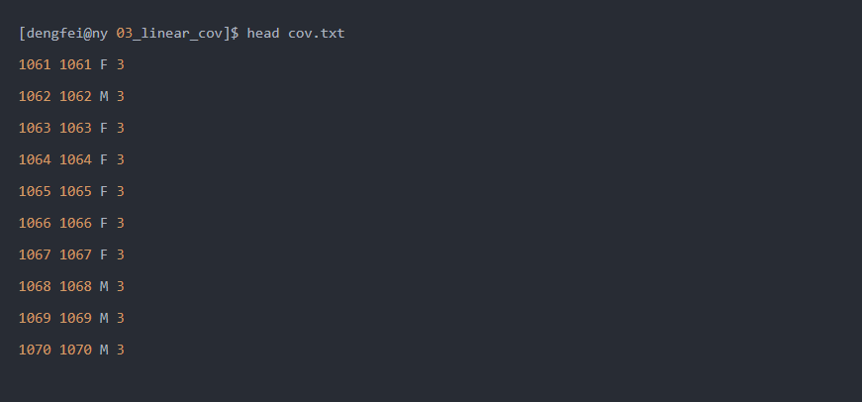
这里第三列为性别,第四列为世代,本篇将世代作为因子,进行因子协变量的GWAS分析。
2.因子协变量
awk '{print $1,$2,$4}' cov.txt >cov1.txt
数据如下:

3.使用plink的dummy coding转化为虚拟变量
plink --file b --covar cov1.txt --write-covar --dummy-coding
结果生成:plink.cov
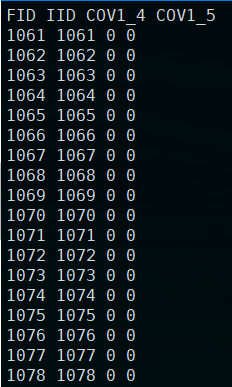
这里的协变量,会减少一个水平,比如本来世代是由3、4、5三个世代,这里只有两个水平。plink文档是这样解释的:
That is, for a variable with K categories, K-1 new dummy variables are created. This new file can be used with –linear and –logistic, and a coefficient for each level would now be estimated for the first covariate (otherwise PLINK would have incorrectly treated the first covariate as an ordinal/ratio measure).
5.进行因子协变量GWAS分析LM模型
代码:
plink --file b --pheno phe.txt --allow-no-sex --linear --covar plink.cov --out re --hide-covar
日志:
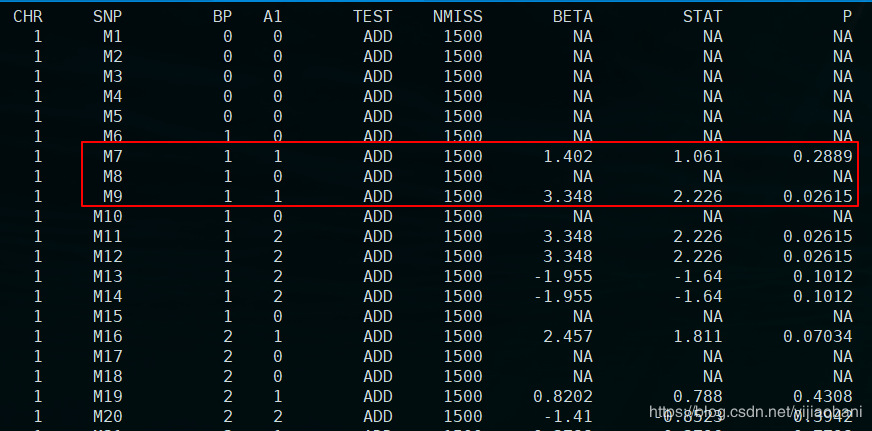
结果文件:re.assoc.linear
6.使用R语言进行结果比较lm+factor
library(data.table)
geno = fread("c.raw")
geno[1:10,1:10]
phe = fread("phe.txt")
cov = fread("cov.txt")
dd = data.frame(phe$V3,cov$V4,geno[,7:20])
head(dd)
str(dd)
mod_M7 = lm(phe.V3 ~ cov.V4 + M7_1,data=dd)
summary(mod_M7)
mod_M9 = lm(phe.V3 ~ cov.V4 + M9_1,data=dd);summary(mod_M9)
M7加上因子协变量结果:
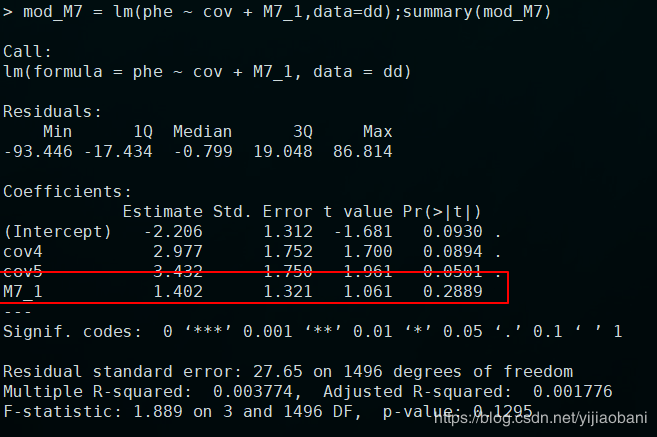
如果是作为数值协变量的结果为:
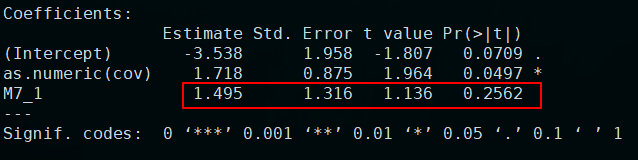
结果是不一样的。
7.使用R语言进行结果比较lm+plink.cov
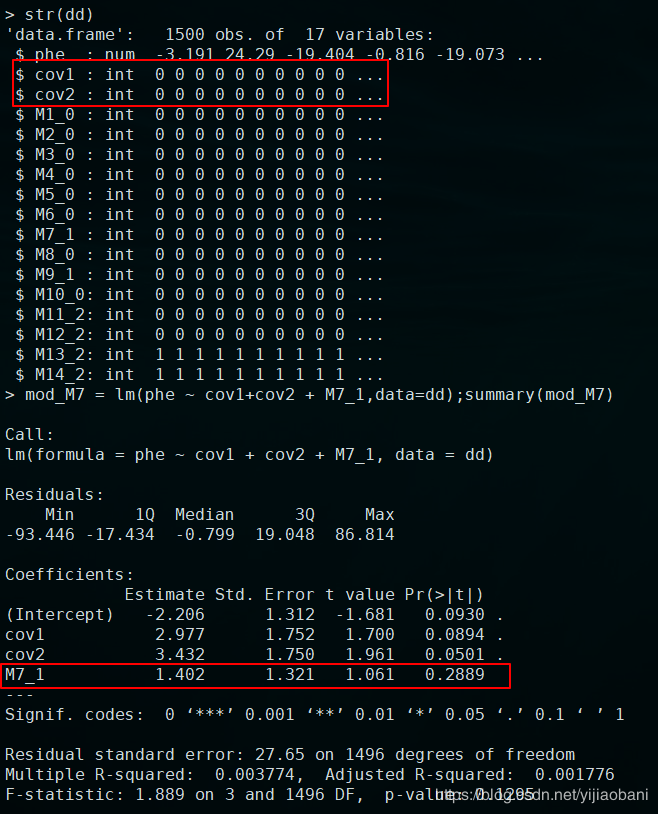
结果和上面世代作为因子完全一样。
8.Note
怎么理解固定即回归这句话的?
R语言中,所谓的因子,在进行回归分析时,也是将其转化为不通过水平的数字变量进行的分析,所以和你手动转化的虚拟变量结果是一样的。