资料来源:公众号:育种数据分析之放飞自我
本篇主要是介绍一般线性模型(LM)中的linear参数,考虑数值+因子协变量,然后将结果与R语言编程结果比较。
GWAS分析时,无论是一般线性模型,还是广义线性模型,都要对协变量进行处理。数值类型的协变量(比如初生重数值协变量,PCA的值)直接加进去,因子协变量(比如不同的年份,不同的地点,场等)需要转化为虚拟变量。
如果一个分析中,既有数字协变量,又有因子协变量,需要将因子协变量转化为虚拟变量后再与数字协变量合并,作为最终的协变量文件进行分析。本次用实际数据进行一下演示。
1.协变量文件整理
第一列为FID ,第二列为ID ,第三列以后为协变量(注意,只能是数值,不能是字符!)
这里协变量文件为:
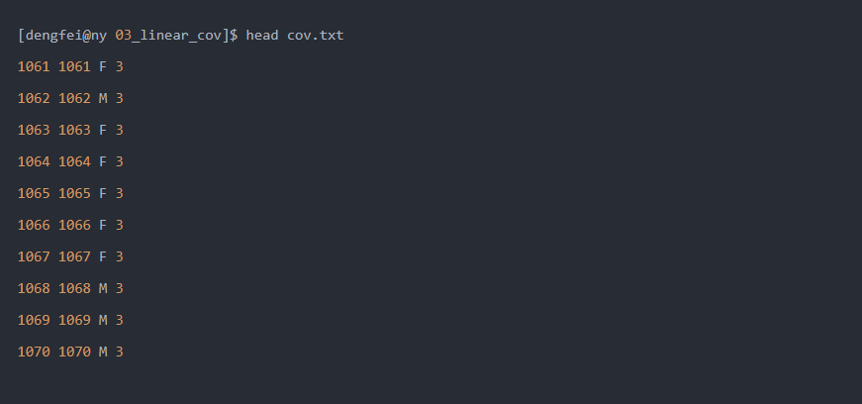
- 这里,首先将F换为1,M换为2,将其转化为连续变量(数字)
- 然后,将世代变为虚拟变量
- 最后,将两个协变量整合到一起
sed 's/F/1/g' cov.txt >cov2.txt
sed -i 's/M/2/g' cov2.txt
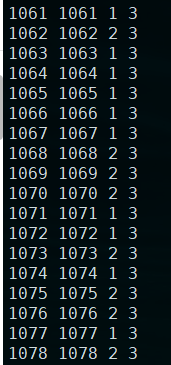
2.使用plink的dummy coding转化为虚拟变量
plink --file b --covar cov2.txt --write-covar --dummy-coding
结果生成:plink.cov
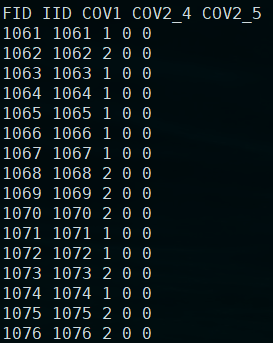
注意:这里的性别虽然是因子,但是其只有两个水平,也可以将作为连续的变量,计算方法是一样的。如果是三个水平的因子,就不能直接转化为变量了。
5.进行协变量GWAS分析LM模型
代码:
plink --file b --pheno phe.txt --allow-no-sex --linear --covar plink.cov --out re --hide-covar
日志:
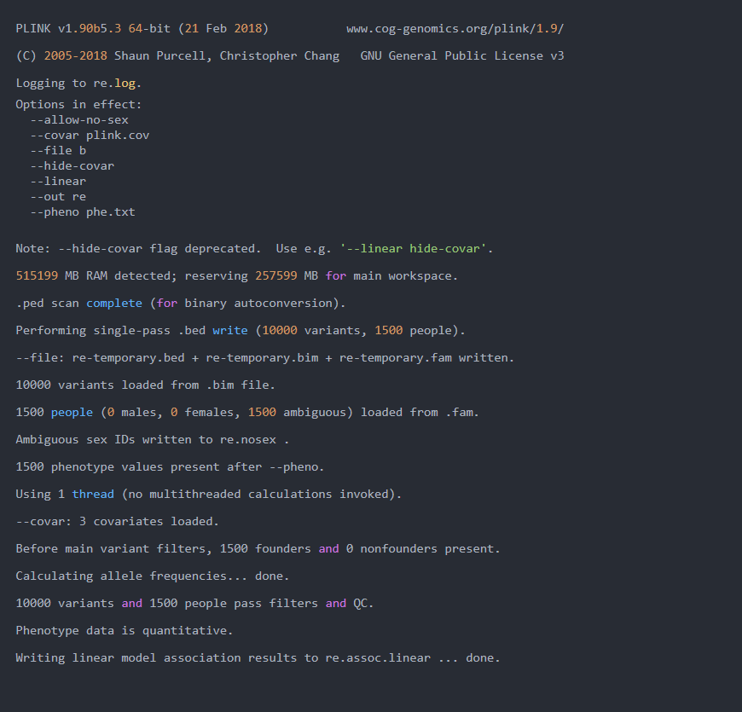
结果文件:re.assoc.linear
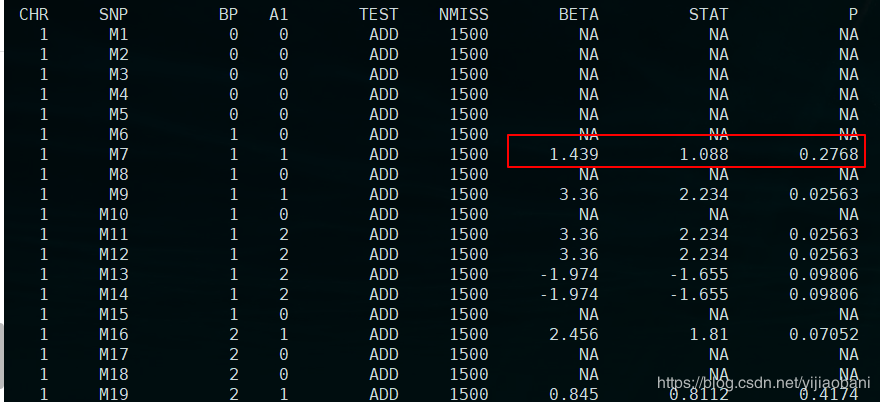
4.使用R语言进行结果比较lm+factor
library(data.table)
geno = fread("c.raw")
geno[1:10,1:10]
phe = fread("phe.txt")
cov = fread("cov.txt")
plink = fread("plink.cov")
dd = data.frame(phe = phe$V3,cov1 = plink$COV1,cov2 = plink$COV2_4,cov3=plink$COV2_5,geno[,7:20])
head(dd)
mod_M7 = lm(phe ~ cov1+cov2+cov3 + M7_1,data=dd);summary(mod_M7)
M7加上因子协变量结果:
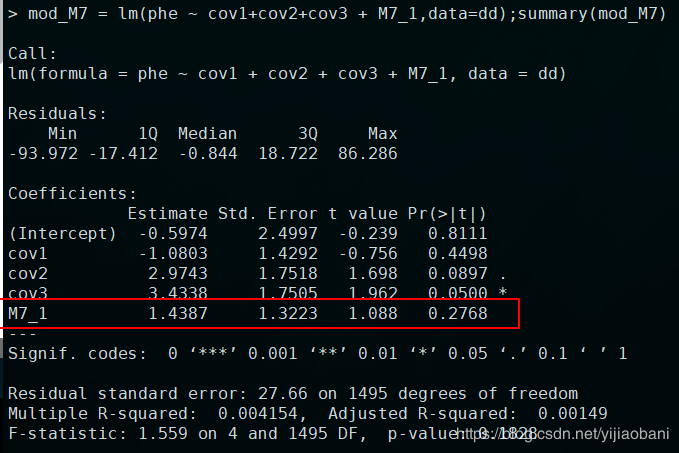
这里,我们可以测试一下:将性别由数字,变为因子,可以发现结果是一样的:
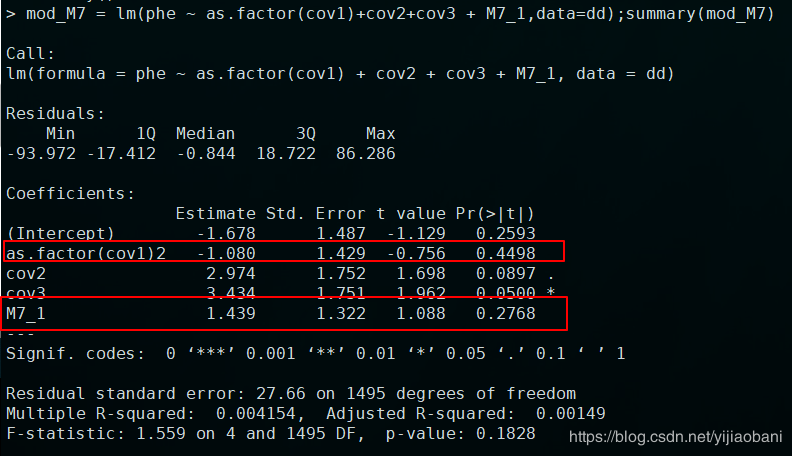
所以当有两个水平的因子(比如性别),变为数字时,对于回归分析而言,两者是一样的结果。