资料来源:公众号:育种数据分析之放飞自我
本篇主要是介绍一般线性模型(LM)中的linear参数,考虑数值+因子+PCA协变量,然后将结果与R语言编程结果比较。
plink做GWAS只有两个模型可以用:GLM和logistic,前者分析数量性状,后者分析二分类性状。而现在GWAS更多使用LMM模型,这个模型plink没法做,以后几篇介绍GEMMA的操作方法。
1.协变量文件整理
第一列为FID,第二列为ID,第三列以后为协变量(注意,只能是数值,不能是字符!)
这里协变量文件为:
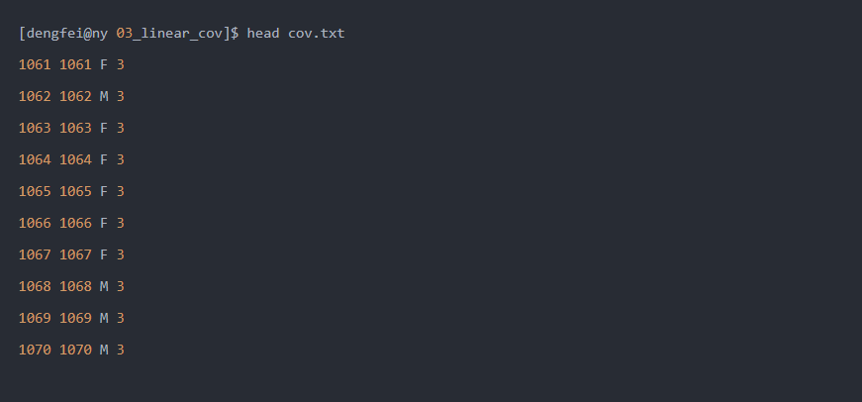
- 首先,将F换为1,M换为2,将其转化为连续变量(数字)
- 然后,将世代变为虚拟变量
- 最后,将两个协变量整合到一起
sed 's/F/1/g' cov.txt >cov2.txt
sed -i 's/M/2/g' cov2.txt
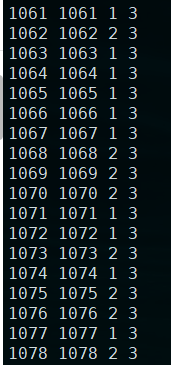
2.使用plink的dummy coding转化为虚拟变量
plink --file b --covar cov2.txt --write-covar --dummy-coding
结果生成:plink.cov
3.使用plink获得pca结果
plink --file b --pca 3
结果文件:
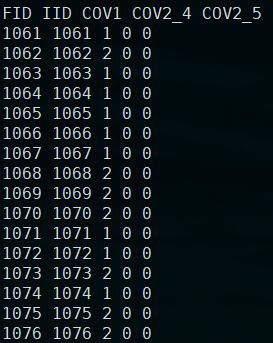
4.将pca结果和协变量结果合并
sed '1d' plink.cov >a.txt
head a.txt
awk '{print $3,$4,$5}' plink.eigenvec >pca.txt
head a.txt
wc -l pca.txt a.txt
paste a.txt pca.txt >pca_cov.txt
合并后的协变量:
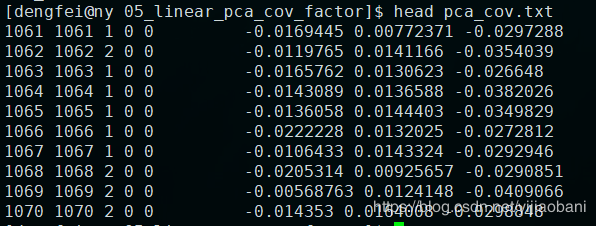
5.进行协变量GWAS分析LM模型
代码:
plink --file b --pheno phe.txt --allow-no-sex --linear --covar pca_cov.txt --out re
日志:
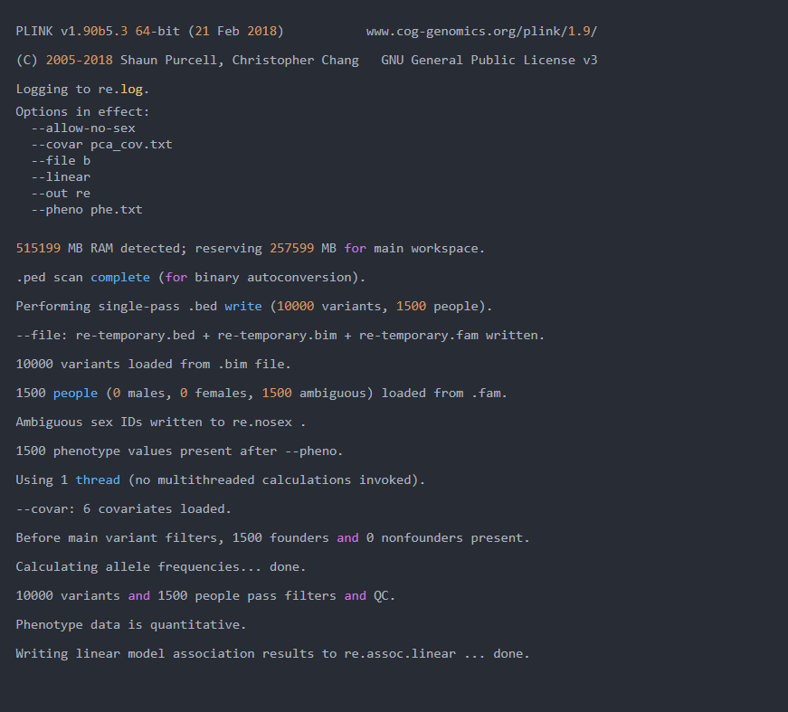
由日志可知,共有六个协变量加入了分析中。
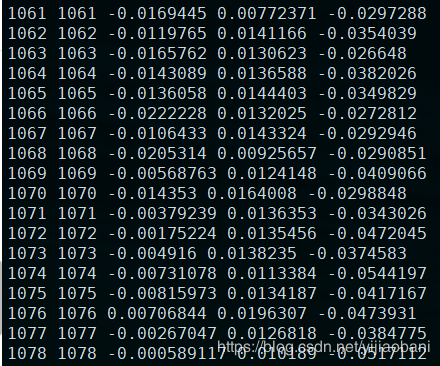
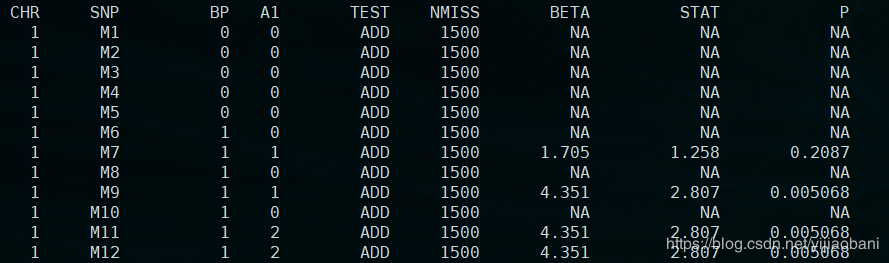
结果文件:re.assoc.linear
6.使用R语言进行结果比较lm+factor+pca
library(data.table)
geno = fread("c.raw")
geno[1:10,1:10]
phe = fread("phe.txt")
plink = fread("pca_cov.txt",header=F,sep=" ")
head(plink)
dd = data.frame(phe = phe$V3,cov1 = plink$V3,cov2 = plink$V4,cov3=plink$V5,pca1 = plink$V6,pca2 = plink$V7,pca3 = plink$V8,geno[,7:20])
head(dd)
mod_M7 = lm(phe ~ cov1+cov2+cov3+pca1+pca2+pca3 + M7_1,data=dd);summary(mod_M7)
M7加上因子协变量结果:
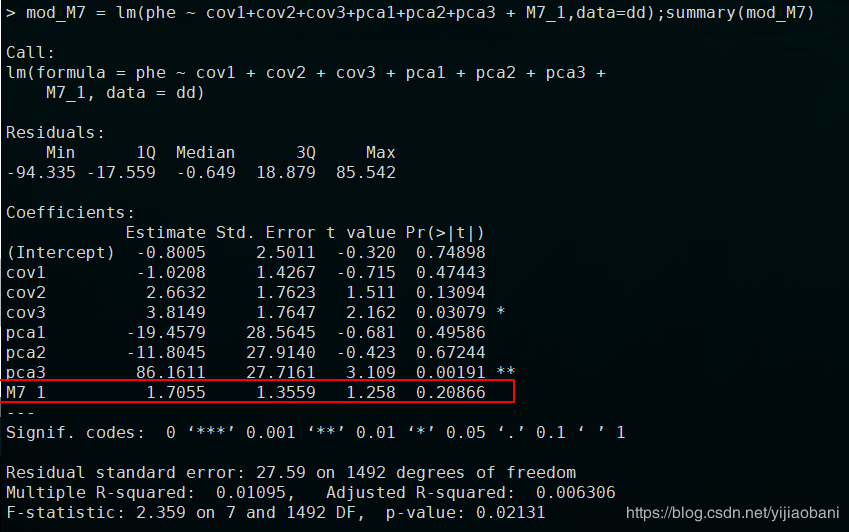
结果完全一样。
7.Note
plink中一般线性模型(LM),linear可以支持数值协变量,因子协变量(经过转化),pca等等,这些过程都可以通过R语言的lm函数复现结果。
一般线性模型可以用plink做,那么混合线性模型怎么做?
gemma!
gemma也可以做一般线性模型,也可以做混合线性模型。plink只可以做一般线性模型,gemma可以利用plink的数据格式做一般线性模型和混合线性模型(但gemma只有linux版本)。