本部分主要参考:[百迈客生物]、[生信菜鸟团]、[Freescience联盟]、[生信草堂]
21.SNP‐level missingness
这是样本中缺少特定 SNP信息的个体数量。具有高度缺失的SNP 可能导致误差。
22.Sex discrepancy(性别差异)
表型信息中的性别与根据基因型确定的性别之间的差异。这个差异可以验证实验室中的样品是否混淆。注意,只有在计算了性染色体(X和Y)上的SNP时,才能进行此测试。
23.关联分析的前世今生
(1)关联分析的定义
关联分析是一种简单、实用的分析技术,即通过数据集中的关联性或相关性研究,描述不同因素之间的规律和模式。
(2)关联分析的方法
在生物学上,一般关联分析是利用统计学方法,基于连锁不平衡原理,将标记与目标性状进行关联,相比于连锁分析,不需要构建特殊的群体且可同时对多个性状进行分析,对QTL定位的精度可达到单基因水平。
(3)全基因组关联分析的提出
其实早在1996年全基因组关联分析的概念就已经被提出,主要是基于基因组范围的高密度标记进行性状的定位研究,但是由于受到基因分型技术的限制,全基因组关联分析在当时并没有得到应用。后来随着高通量测序技术的诞生,直到2005年以来 GWAS(全基因组关联分析)才开始普遍应用起来。GWAS克服了候选基因关联的局限性,许多位于基因间的变异以及存在于功能尚不明确的基因中的SNP都可以被定位出来,并且大部分变异是之前的研究中未被发现的,这就为性状遗传机理的全面揭示提供了可能性。当然,传统的GWAS分析也存在一定的局限性:
- 通常GWAS适用于常见变异研究,对稀有变异检测不敏感;
- 大多数显著的SNP位点并非真正影响目标性状的功能变异,而是与其处于连锁状态;
- 通过不同的模型及阈值筛选,会有假阳性或假阴性的SNP产生;
- 目前主流的GWAS分析方法仍然是基于SNP的关联,会有一些重要的其它突变类型丢失,即所谓的遗传力的丢失;
- 同时基因互作的效应也无法检测,因此需要对GWAS进行升级和优化去解决这些问题。
24.GWAS简介
24.1 什么是GWAS?
全基因组关联分析(Genome-wide association study;GWAS)是应用基因组中数以百万计的单核苷酸多态性(single nucleotide ploymorphism,SNP)为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,通过比较发现影响复杂性状的基因变异的一种新策略。
GWAS是在某一特定人群中研究遗传突变和表型之间的相关性。GWAS的理论基础是连锁不平衡定律(linkage disequilibrium, LD),既假设观察到的SNP与真正的致病突变(causal variant)之间存在很强的LD。基于基因芯片设计的GWAS目前着重关注人群中的常见变异(common SNPs, 通常指最小等位基因频率MAF > 0.01),因此,通过GWAS发现的疾病易感位点主要集中在常见变异上,也即是通常所说的common disease,common variants理论。GWAS的概念是在群体遗传学的概念下发展起来的,其统计效应受到样本量的直接影响。Am J Hum Genet文章的观点认为能检测到显著关联结果的最小的样本量是基因分型(SNP array plus imputation or WGS),allele frequency 以及effect size的函数。
24.2 通过GWAS,我们发现了什么?
(1)GWAS找到了性状的易感基因
大规模的GWAS研究找到了大量显著的遗传变异与表型的相关,涉及到的表型包括:常见疾病,数量性状,社会行为性状(如幸福感等),甚至是基因表达和DNA甲基化水平。这些显著影响性状的遗传变异中,有一部分已经指导了分子实验室的工作去发掘潜在的治病机理,比如FTO和肥胖,MHC和精神分裂症。
(2)多基因型效应
当我们更加深入地去看复杂性状的整个遗传构成,又会得出一些重要的结论。最开始的观察还是来自与最显著的位点,科学家们发现这些达到统计阈值的遗传变异也仅仅只能解释很少一部分的表型差异。根据此现象,澳大利亚昆斯兰大学的华人学者杨剑认为GWAS阈值以下的常见变异也能贡献相当一部分的遗传力。因此,他们团队在最近发表在Am J Hum Genet的综述中指出复杂性状的多基因特点。比如,2008年的时候,GWAS找到了40个显著控制身高的SNP,但这些SNP仅仅解释了大约5%的遗传力。到了2014年,在整个基因组上有超过700个遗传位点被认为与身高有关,它们贡献的遗传变异达到20%以上。
(3)从多基因型到泛基因型
今年6月份发表在CELL上的一篇前瞻性文章真可谓掀起了了GWAS研究的轩然大波。通过探究GWAS信号在基因组上的分布情况,研究人员发现对于身高这样的复杂性状,其作用位点广泛地分布在整个基因组上面,意味着几乎全部的基因都参与到对身高的调控。更重要地,cell/tissue specific表达的基因在控制性状过程中起到决定性的作用。通过将GWAS信号富集到不同细胞中活跃的染色质区域,他们发现精神分裂症的遗传贡献主要来自与神经系统相关的基因,比如铁离子通道和钙铁转运等。
24.3 GWAS还能做什么?
学科的发展决不是哪一块是热点就做哪一块,GWAS也一样,总是成果和质疑并存。质疑的声音存在是因为取得的成果还不足以回答所有的科学问题。那么,GWAS的未来就应该朝着那些暂未解决的问题前进。
(1)复杂性状的差异到底由多少遗传位点或基因决定?
由于imperfect LD的存在,我们不能假定观察到的SNP能够tag所有可能的效应位点。随着全基因组测序技术的广泛应用和成本降低,越来越多的GWAS研究用测序取代了基因芯片,从而有机会覆盖到基因组上的全部遗传标记。由于样本量的制约,每一个有微小效应的SNP不一定能够达到统计阈值。随着世界各地大规模人群队列的建立,大量的样本积累可以显著地增加GWAS的统计效应。因此,未来的GWAS研究将会收集数以百万的人群样本,采用全基因组测序的手段探究性状的遗传结构。
(2)具有显著效应的位点以怎样的方式调控表型?
揭示复杂性状背后的分子机制的主要挑战在于整合多组学数据进行联合分析。特别是在GWAS找到的位点大多数位于非编码区的情况下,探索细胞内分子调控网络显得尤为重要。研究表明,通过整合基因表达和DNA甲基化数据,多数位于非编码区的GWAS信号可以调控特定细胞和组织中的基因表达和DNA甲基化水平。
(3)如何研究低频变异?
前面提到,基于基因芯片的GWAS无法检测到低频变异的遗传贡献,而现在的理论是低频变异往往具有更大的遗传效应。低频变异的研究方法一般是burden test,即假设多个独立的效应位点集中在一个基因区域内起作用。但该方法需要根据遗传标记的功能和频率判断该位点是否可以被纳入burden test。
(4)从GWAS能否到性状预测?
早在2007年,就有观点认为GWAS数据能够作为疾病或复杂性状的遗传预测。目前遗传预测的关键制约因素是没有足够大的样本获得足够精确的多基因风险分数(polygenic risk score)。多基因风险分数并不适用于单个个体,其主要的作用在于区分出最高和最低风险的组。因此,GWAS真正要服务于临床或者为近年来火热的“精准医学”提供理论依据还有很长的路要走。
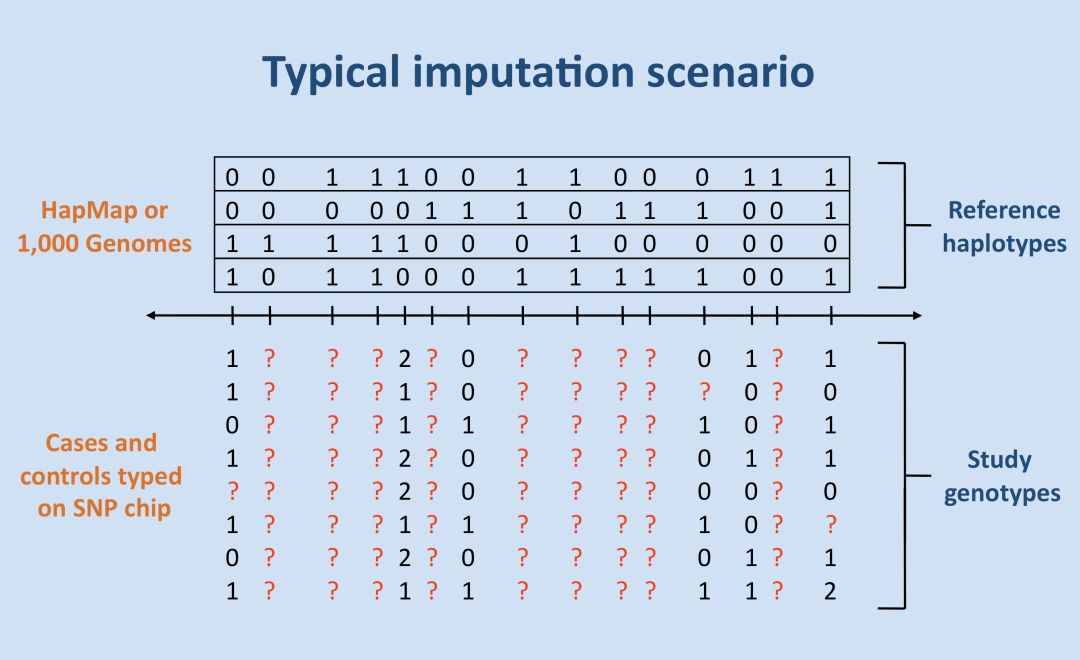
25.基因型填充(genotype imputation)
相比全基因组测序,GWAS芯片确实更加经济,但是其缺点也是显而易见的,只能够分析挖掘已知的SNP位点,而且位点数据量相对较少,要知道一个全基因组测序分析得到的SNP位点在几百M左右。为了解决这个问题,科学家提出了基因型填充的思想。
基因型填充(genotype imputation)是全基因组关联分析(genome-wide association study, GWAS)中的重要工具,通过这项技术可以精确地预测没有被芯片设计所覆盖的多态性位点的基因型,使得更多的遗传位点应用到关联分析中,从而提高发现新的致病基因的可能性。
首基因型填充需要由高密度SNP构成的单体型(haplotypes)作参考模板,根据样本已有的分型结果,与参照的单倍型进行比较,确定其可能所属的单倍型,然后进行填充。
随着千人基因组计划的完成,超过7千万的多态性位点被发现,由此构建了一张丰富的人类遗传单体型图谱,为基因型填充提供了有力依据。借助高性能计算的快速发展,越来越多的研究人员选择利用基因型填充对芯片产生的基因型数据进行imputation,进而增加GWAS和fine-mapping的效能。
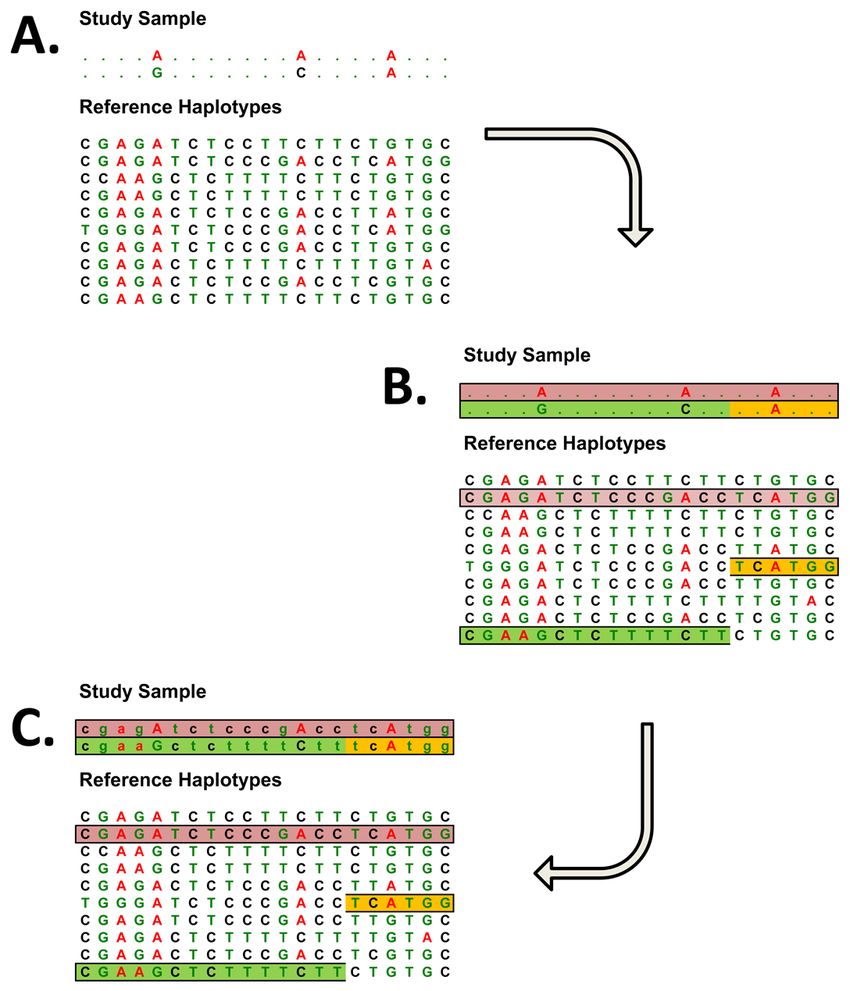
从以上示意图可以看出,基因型填充有两个必要条件,第一个条件就是参照的单倍型,对于独立样本,可以采用hapmap或者1000G等项目的单倍型作为参照,第二个条件就是已知分型结果位点的比例和分布,对于需要填充的样本,要保证一定密度的分型结果,需要根据已有的分型结果来推断该样本可能的单倍型,分型结果已知的位点越多,其单倍型推断的准确性越高,填充的准确性才会越高,根据这个条件来看,GWAS芯片最适合进行基因型填充,因为其覆盖的SNP位点的数量和分布更有助于推测样本的单倍型。
Imputation的软件:
- Beagle
- IMPUTE2
- MACH
26.使用IMPUTE2进行基因型填充
IMPUTE2是一款基因型填充软件,和其他软件相比,其填充的准确率最高。其提供了以下两大功能:
- Haplotype phasing:单倍型分析
- Genotype imputation:基因型填充
27.基因型填充中的phasing
phasing其实就是haplotype estimation的别名
目前主流的基因型填充软件都分为了以下两个步骤
- Phasing genotypes
- Imputing ungenotyped markers
这个二步法是为了提高运算速度而设计的,基因型填充有一步法和二步法两种策略,示意如下
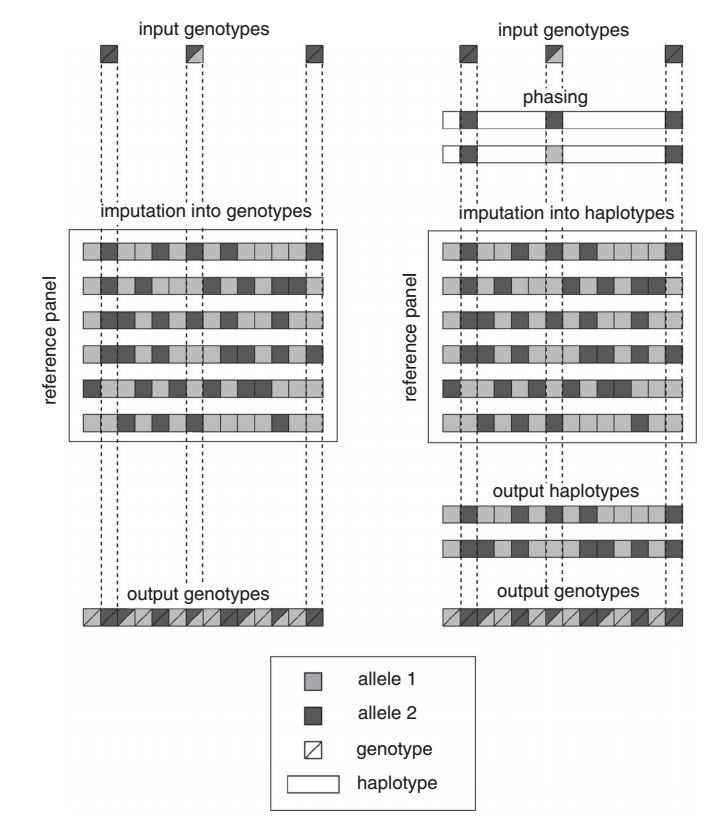
一步法根据refernce panel的基因型结果推测样本可能的单倍型构成,然后直接填充缺失的基因型;二步法首先根据已有的分型结果推测样本对应的单倍型,然后用单倍型与reference panel的单倍型进行比较。
reference panel中SNP的位点数量是巨大的,当reference panel的版本更新时,就需要重新推断一遍样本的单倍型,而且根据数量巨大的reference panel的分型结果进行推断,运算量巨大,二步法很多的解决了这个问题,直接根据样本的分型结果来推断基因型,和reference panel无关,所有样本只需执行一次即可,用单倍型和reference panel进行比较,运算量也相对减少,所以二步法的运算速度更快。
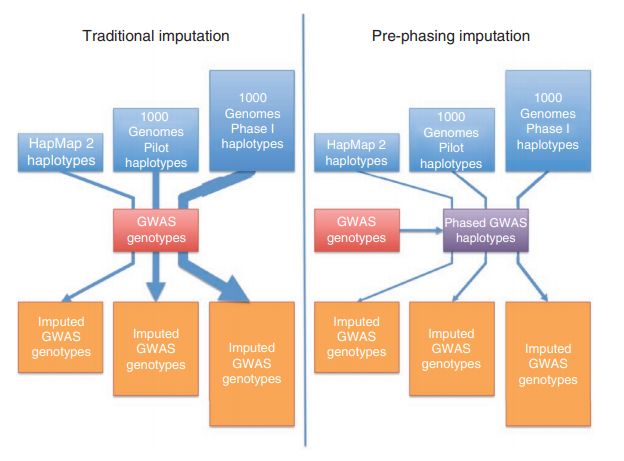
Fast and accurate genotype imputation in genome-wide association studies through pre-phasing
左侧为一步法,根据不同的reference panel运算3次,右侧为二步法,首先得到phased haplotypes, 然后只需要和不同版本的reference panel比较即可,运算量大大降低。基于phasing的二步法有效提高了基因型填充的运行效率,使得基于大规模reference panel的基因型填充得到更广泛的应用。
Phasing的软件:
- MaCH
- PHASE
- fastPHASE
- PL-EM
- BEAGLE
- SHAPEIT
28.使用SHAPEIT进行单倍型分析(phasing)
phasing其实就是haplotype estimation的别名
SHAPEIT是一款单倍型分析工具,运算速度快,准确率高,是impute2官方推荐的pre-phasing工具,我们实验室使用的也是该款软件:
29.Q-Q plot图的介绍
Q-Q plot是关联分析结果可视化的一种经典方案,这里的Q代表quantile, 分位数的意思,关联分析的Q-Q plot示意如下:
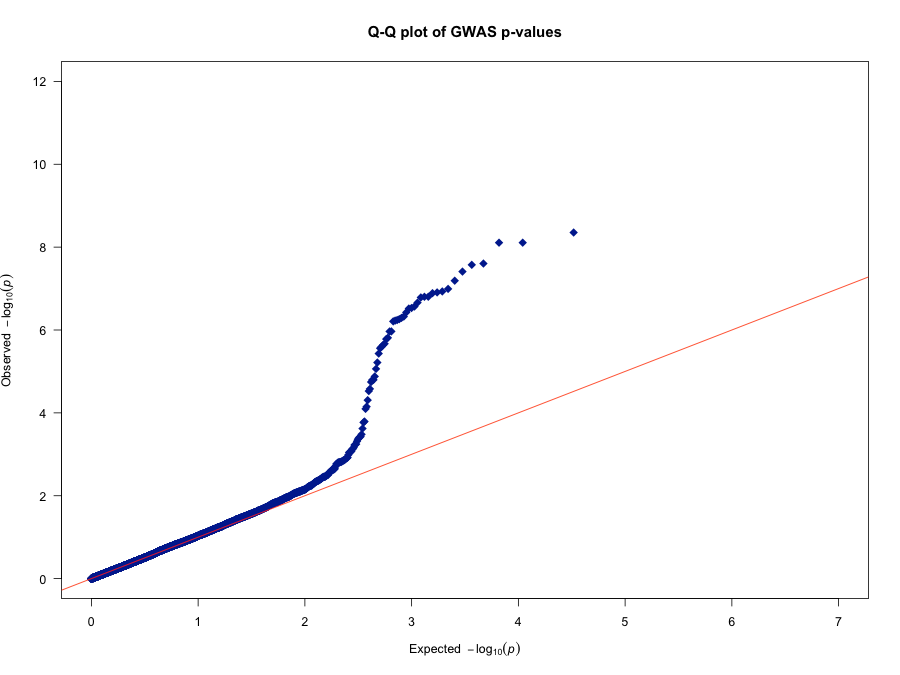
x轴代表期望p值,y轴代表实际p值。
30.曼哈顿图的介绍
作为一种经典的可视化方式,曼哈顿图使用广泛,在GWAS分析中随处可见。曼哈顿图的命名得益于其形状:

GWAS中的曼哈顿图示意如下:
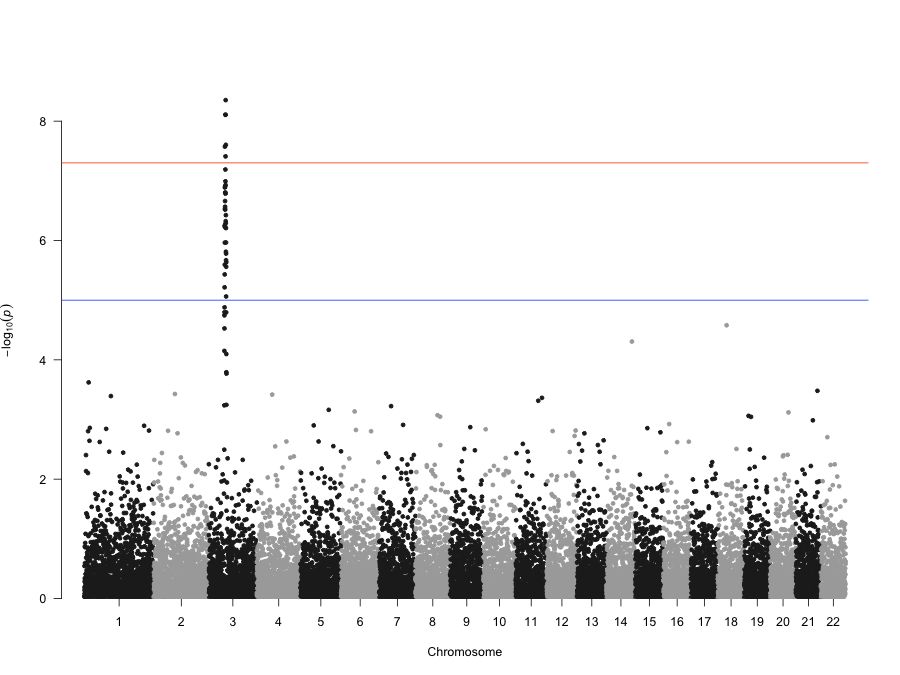
曼哈顿图的x轴为snp位点在染色体上的位置,y轴为SNP位点对应的p值。
31.FUMA软件——对SNP位点进行功能注释
通过GWAS分析可以找出与疾病相关联的SNP位点,然而我们的根本目的是找出可能导致疾病发生的SNP位点,这些位点位于GWAS分析结果中,完成了GWAS分析之后,我们还需要借助下游分析对GWAS的分析结果进一步注释和挖掘,筛选可能致病的SNP位点。
相关的注释网站和数据库很多,为了更加方便和有效的利用各种资源,科学家搭建了一个整合型的在线网站FUMA,通过该网站,可以对GWAS分析结果中的SNP位点进行功能注释,基因优先级排序,并提供了结果的交互式展示,共包含以下两个模块:
- (1)SNP2GENE, 该模块输入GWAS分析的结果,对其中的显著SNP位点进行功能注释
- (2)GENE2FUNC, 该模块输入为基因列表,对基因的功能进行注释
32.GWAS中的荟萃分析(Meta-analysis)
对于GWAS分析而言,增加样本量是提高检验效能的最直接有效的方式。目前常规GWAS项目的样本量约为1000 cases vs 1000 controls,这样的样本量能够检测到的相关SNP位点基本属于common SNP, 频率在1%以上,对应的OR值也通常在1.2以上,对于低频和罕见突变位点,常规的样本量则无法有效检出,因为携带对应Allel的样本太少,很难达到统计学显著性。
对于多基因复杂疾病而言,其相关联的的SNP位点肯定不仅限于common SNP, 为了有效检测相关联的低频和罕见变异位点,需要进一步增加GWAS分析的样本量,然而考虑到样本收集的难度,周期和实验成本,单个项目很难达到有效的样本量。鉴于这个情况,最直接的解决方案就是合并多个GWAS项目的结果,来达到增加样本量的目的。多个数据集的合并分析,正是meta分析大展身手的时候。
meta-analysis, 称之为元分析,或者荟萃分析,早在1976年就提出了这个概念,其分析对象是已有的研究成果,用来对先前研究进行综合评价和定量合并,在多个领域都有其应用。在生命科学领域,包括基因组,转录组,蛋白质组等多组组学研究领域,都有meta分析的用武之地,而GWAS的meta 分析就是meta分析在基因组领域的最经典应用场景。
meta分析有多种算法,比如基于pvalue, 基于排序,基于效果量等各种算法,每种算法各有优劣。时至今日,相关的工具和软件也很多,对于GWAS的meta分析而言,METAL、PLINK、MetaSoft等软件都可以实现对应的功能。
在整合多个数据集的分析结果时,我们需要考虑到不同数据集之间的差异,也称之为数据集的异质性,常用的有以下两种模型:
- Fixed effects models, FEM
- Random effects models, REM
第一种称之为固定效应模型,第二种称之为随机效应模型,固定效应模型中假设不同数据集的差异是固定,而随机效应模型假设不同数据集的差异服从正态分布,因此,固定效应模型适用于相同实验条件下的数据,而随机效应模型则可以用于处理不同来源的独立数据。
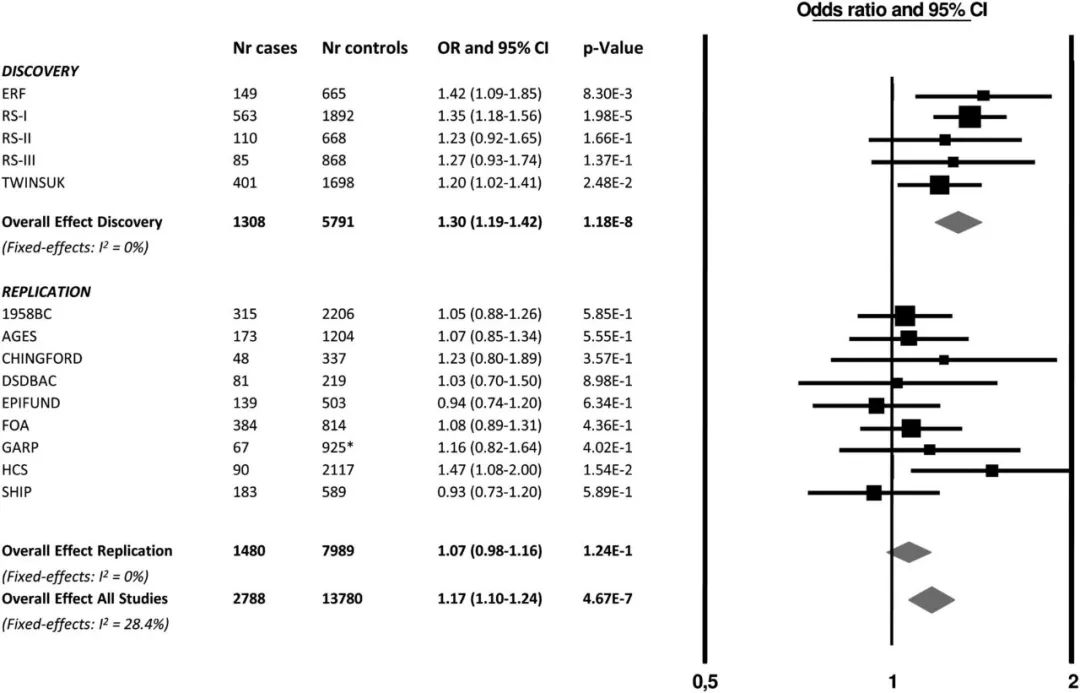
对于GWAS meta分析的结果,经典的可视化方式是forest plot(森林图), 其展示了每个study中变异位点对应的OR和P值:
33.GWAS中用于meta分析的软件——METAL
METAL是gwas meta分析最常用的工具之一,该软件支持以下两种meta分析的算法:
- pvalue
- standard error
34.LDSC:连锁不平衡回归分析
通过GWAS分析可以识别到与表型相关的SNP位点,然而严格来讲这个结果并不一定真实客观的描述遗传因素对表型的效应,因为其结果是由以下两个因素共同构成的
- (1)polygenic effects, 基因对表型的效应
- (2)confounding factors, 混淆因素,比如群落分层,样本间隐藏的亲缘关系等等
尽管我们在GWAS分析中,可以通过协变量来校正群落分层等因素,但是混淆因素是无法完全消除的。为了保证分析结果的准确性,我们就需要评估GWAS分析结果中以上两个因素的占比,只有当混淆因素占比很低时,才能说明我们的分析结果是可靠的,此时我们就可以通过LDSC来探究这个混淆因素的占比。
35.Inbreeding coefficient近交系数的计算
inbreeding coefficient,中文翻译为近交系数,近婚系数,近亲交配系数等等,用大写字母F表示。要理解这个概念,首先要搞清楚”近亲”的定义。
近亲指的是三代及以内的具有共同血缘关系的个体,他们之间的婚配称之为近亲结婚。由于双方包含了很多相似的遗传因子,其后代个体的纯合基因的比例会增加,患常染色体隐性遗传病的风险也会急剧增加。
从基因层面来说,近亲婚配的后果就是一个基因的allele来自共同祖先, 即血缘同源IBD。为了更加客观的描述个体间近亲婚配情况,提出了以下两个概念:
- (1)coffcient of relationship, 针对两个个体间,表示的是两个个体间来自共同祖先的同源基因比例, 称之为共祖系数
- (2)cofficient of inbreeding, 针对一个个体,表示的是该个体任意一个基因的两个allele来自同一个祖先的概率,称之为近交系数
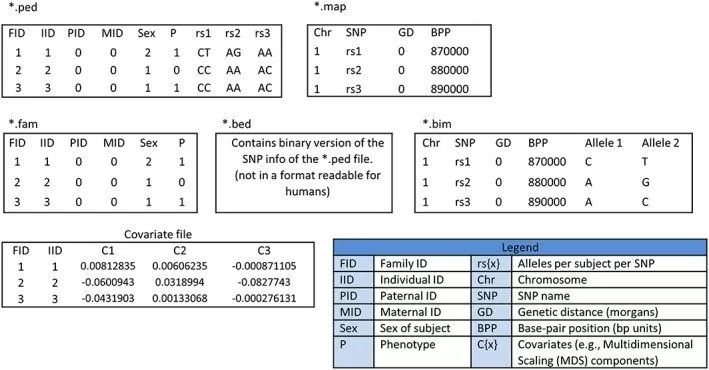
36.各种常用PLINK文件概述
PLINK 既可以读取文本格式的文件也可以读取二进制文件。读取大型文本文件时,建议使用二进制文件。
(1)文本格式的 PLINK 数据包含两个文件:
- 包含相关个体及其基因型的信息(*.ped);
- 包含遗传标记的信息(*.map)。
(2)二进制 PLINK 数据由三个文件组成:
- 包含样本标识符(ID)和基因型(*.bed)的二进制文件;
- 包含相关个体信息文件(*.fam);
- 遗传标记文件( *.bim)。
例如,我们要进行一项躁郁症研究,.bed 文件将包含所有患者和健康对照组的基因分型结果;.fam 文件将包含与受试者相关的数据(与研究中其他参与者的家庭关系,性别和临床诊断信息);*.bim 文件将包含 SNP 物理位置的信息。使用协变量进行分析通常需要第四个文件,其中包含每个个体的协变量值。
37.使用PLINK进行遗传数据的质控
(1)SNP和个体水平的缺失
- –geno:排除大部分受试者中缺失的SNP。
- ‐‐mind:排除基因型缺失率高的个体。
建议首先基于宽松的阈值(0.2;>20%)过滤 SNP 和个体,过滤掉丢失程度很高的SNP和个体。然后再应用更严格的过滤参数(0.02)。注意,应先执行SNP过滤再进行个体水平过滤。
(2)性别差异
- ‐‐check‐sex:根据X染色体杂合/纯合率检查数据集中记录的性别与真实性别之间的差异。
如果许多样本存在此类差异,则应仔细检查数据。男性X染色体纯合度估计值>0.8,女性<0.2 。
(3)次要等位基因频率(MAF)
- ‐‐maf:仅输出高于MAF阈值的SNP。
MAF低的SNP很罕见,因此缺乏检测 SNP 表型关联的power。这些SNP也更容易出现基因分型错误。MAF阈值应取决于样本量,较大的样本可以使用较低的 MAF 阈值。对于大样本(N=100,000)与中等样本(N= 10000),通常将0.01和0.05用作MAF阈值。
(4)哈迪-温伯格平衡(HWE)
- ‐‐hwe:过滤偏离 Hardy-Weinberg 平衡的标记。
这是基因分型错误的常见指标,它也可能指示进化选择。对于二元性状,建议:HWE p值<1e-10(病例)和 <1e-6(对照)。不太严格的病例阈值可避免丢弃选择中的疾病相关SNP。对于数量性状,建议HWE p值<1e-6。
(5)杂合率
- 排除杂合率高或低的个体。
此类偏差可能表明样品受到污染或近亲繁殖。建议删除样本杂合率均值偏离±3 SD的个体。
(6)相关性
- ‐‐genome:计算两两样本之间的血缘同一性。使用独立的 SNP(pruning)进行此分析,并将其限制为仅常染色体。
- ‐‐min:设置阈值并输出关联性高于所选阈值的样本列表。可以检测到与 pihat> 0.2 相关的个体(即,二级亲戚)。
这些相关性可能会干扰关联分析。如果你有一个基于家庭的数据集(例如,父母后代),则不需要删除相关对,但统计分析应考虑家庭之间的相关性。但是,对于基于人口的样本,建议使用pihat阈值为0.2。
(7)群体分层
- ‐‐genome:计算两两样本之间的血缘同一性。使用独立的 SNP(pruning)进行此分析,并将其限制为仅常染色体。
- ‐‐cluster ‐‐mds‐plot k:根据 IBS ,生成数据的 k 维子结构。k 是维度(通常为10)。这是 QC 的重要步骤,由多个程序组成。此步骤将在“控制群体分层”一节中详细描述。
通常,如果样本包含多个种族群体(例如,非洲人,亚洲人和欧洲人),建议分别对每个种族群体进行关联测试,并使用适当的方法,例如荟萃分析,以合并结果。如果你的样本仅来自单个种族,则可以通过下面介绍的方法校正群体分层。
38.使用PLINK控制群体分层
如上所述,群体分层是GWAS系统误差的重要来源。研究表明,在单个种族中也可能存在一些群体分层的现象。因此,检测和控制群体分层是至关重要的QC步骤。
有多种方法可以校正群体分层。本部分将介绍PLINK中包含的一种方法:多维缩放(MDS)。该方法计算个体之间共享的全基因组等位基因平均比例,以生成每个个体遗传变异的数量指标(组分)。我们可以绘制各个组成部分的分数来探索这些样本是否存在在遗传上彼此相较预期更相似。例如,在一项包括来自亚洲和欧洲的受试者的研究中,MDS 分析表明,亚洲人彼此之间在遗传上比欧洲人更相似。
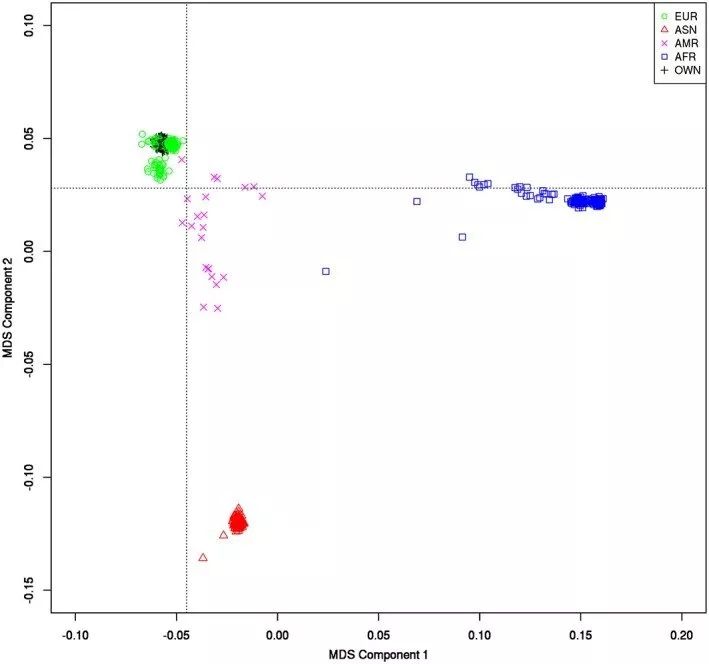
图3 举例说明了这种分析。
基于MDS分析的异常个体应在进一步的分析中剔除。在排除了这些个体之后,必须进行新的MDS分析,并且将其主成分用作关联分析的协变量,以校正群体内的群体分层。需要包括多少成分取决于人口结构和样本量,但是精神遗传学界普遍接受最多包含10个成分。
39.使用PLINK进行二元/数量性状的关联统计检验
经过QC和MDS成分计算后,数据已准备就绪,可用于后续关联检验。
(1)二元性状计算
在PLINK中,SNP与二元性状的关联(1=不受影响,2=影响; 0和-9表示缺失;这些为 PLINK 中的默认选项,可更改 )可通过 ‐‐assoc 或 ‐‐logistic 进行分析。
- ‐‐assoc参数将进行卡方关联检验,不允许引入协变量。
- ‐‐logistic参数将执行logistic回归分析,该分析允许包含协变量。
- ‐‐logistic 比 ‐‐assoc 更加灵活,但也会增加计算时间。
(2)数量性状计算
在PLINK中,可使用 ‐‐assoc 和 ‐‐linear 来计算SNP与数量性状的关联。
- 当PLINK检测到数量性状( 即1、2、0或其他值 )时, ‐‐assoc 将通过执行 Student’s t 检验的渐近版本以比较两个均值来,不允许引入协变量。
- ‐‐linear 则以每个单独的SNP作为预测变量执行线性回归分析。
- 与 ‐‐logistic 参数类似,‐‐linear 可以引入协变量,但比 ‐‐assoc 要慢一些。
(3)多重检验校正
基因分型芯片可以同时进行多达400万个标记的基因分型,从而产生大量的统计检验,产生多重检验负担。基因型填充也可能会进一步增加检验关联的数量。各种模拟表明,无论研究的实际SNP密度如何,用于我们广泛使用的全基因组显著性阈值5×10^-8足以控制整个基因组中独立SNP的数量。在测试非洲人口时,由于这些个体之间更大的遗传多样性(可能接近 1.0×10^-8 ),因此需要更严格的阈值。
Bonferroni校正,Benjamini-Hochberg错误发现率(FDR)和置换检验是三种广泛使用的确定全基因组显著性的方法。
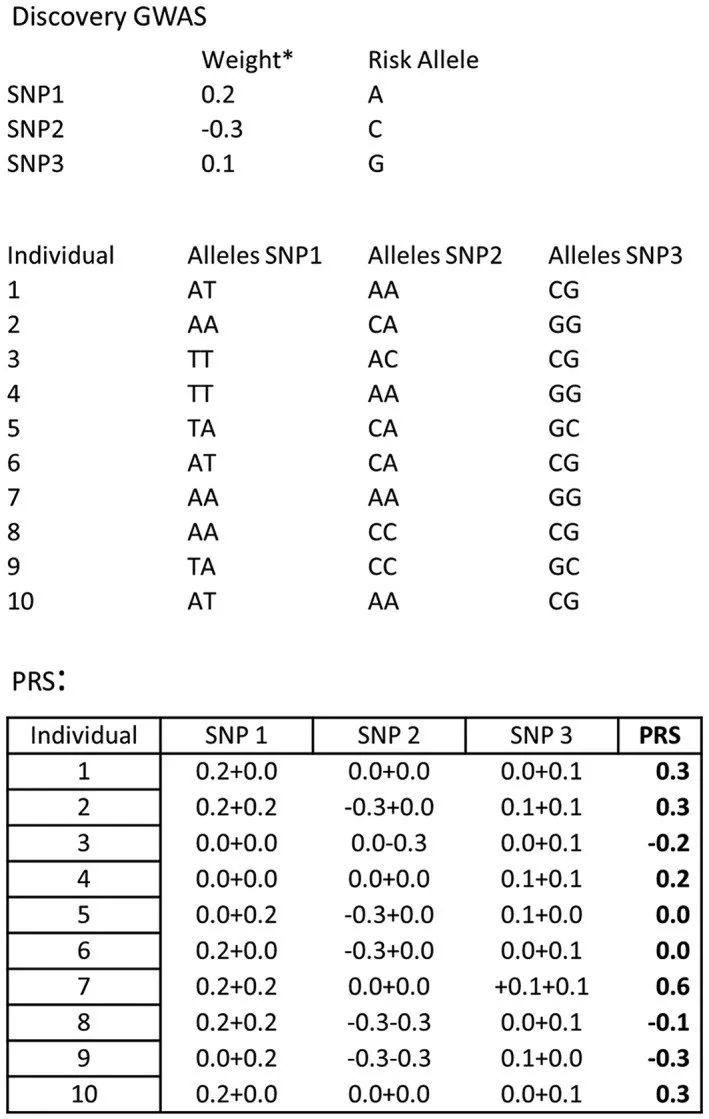
40.多基因风险预测分析(PRS分析)
将三个单核苷酸多态性(SNP)汇总为一个多基因风险评分(PRS)的例子。权重可以是 beta 或比值比的对数,具体取决于分析的是数量性状还是二元性状。