本部分主要参考:[百迈客生物]、[生信菜鸟团]、[presentlife]、[知乎-ffa]、[橙子牛奶糖]、[赵煮机]
41.GWAS相关的四个数据库
基于GWAS研究,已有很多公共资源使我们受益良多。
(1)GWAS ATLAS
GWAS ATLAS数据库收录了来自4756个人类不同表型的GWAS结果,并进行了不同表型间的遗传相关性分析,对应的文献发表在nature genetics上。该数据库不仅仅是一个gwas结果存储的数据库,对于收录的原始gwas结果,进行了深入挖掘,提供了risk loci, LDSC, MAGMA基因集关联分析,多种表型间的遗传相关性分析等结果。
(2)GWAS Catalog
GWAS Catalog是EBI旗下的一个数据库,收录了公开发表的GWAS分析结果,GWAS Catalog详细介绍。
此外,从GTEx中我们可以获得SNP与基因表达之间的关联信息。Ensembl和FUMA是功能注释的常用工具。我们还可以免费获得对所研究的精神病性状或疾病的遗传结构产生重要见解的研究方法。例如,GCTA和 LD评分回归分析已用于估计基于SNP的遗传力。
(3)GTEx Portal
查看基因表达的网站:https://www.gtexportal.org/
(4)全基因功能注释HiChIP数据库
HiChIP数据库具有丰富的可视化功能,首页即可通过人体器官的交互式动态展示页面快速地选择对应的细胞类型数据进行访问。在浏览页面,HiChIP相互作用数据以“器官-组织-细胞系”的树状结构进行呈现,还有可交互式的饼状图来提供不同器官或者组织下HiChIP相互作用数据的数量分布。http://health.tsinghua.edu.cn/hichipdb/
42.GWAS关联区间
确定GWAS关联区间一般有两种方法,一种是peak SNP上下游200kb,这个方法简单粗暴,每个位点的区间也是固定不变。一种是依赖于LD block,由于染色体不同位置的LD不同,所以这种方法区间大小都不一致,得到的区间大小也更准确。
43.性状和QTL
凡是肉眼可见的表现都能称之为性状(当然还有肉眼凡胎看不到的性状)。在某一个物种中,某个性状可能会有不同的表现,比如人头发有直有卷,孟德尔的豌豆有圆有扁。这种我们称之为相对性状。性状由对应的基因控制。
根据控制基因的个数,可以把性状分为质量性状和数量性状。质量性状由一对或者少数几对基因控制,表现在表型数据上就是不连续的变异,比如人的性别。数量性状是连续变异的性状,其遗传基础复杂,受多基因控制,且易受环境影响,比如人的身高。
通常大多数的性状为数量性状,控制数量性状的基因在基因组中的位置称为数量性状基因座即QTL(quantitative trait locus)。
44.GWAS的困境和遗传模型的新思
表型和基因型的游戏、不够“翘”的QQ图、哪里出问题了?、“开个天窗”的解释、拆了房子吧、表型的思考
很有意思的文章,链接点此。
昝老师对该文的一点纠正:遗传率是在方差水平上的一个概念,而文中说的是均值的概念。确切的例子可以是,一群人高矮不一,他们的身高方差是Vp,那么方差Vp的80%是由于遗传造成的
45.GWAS模型
群体结构(Q矩阵或主成分分析)全基因组关联分析的理论基础是SNP标记与目的基因关联(LD),这里的SNP只是作为标记,本身是否有生物学意义还不知道。研究的群体可能内部还存在分层,有一些基因的频率在不同亚群之间就是不一样,LD也不相同。这些都需要校正。考虑了群体结构的模型称为一般线性模型(GLM)。
个体间亲缘关系矩阵(Kinship)用来校正群体内个体间复杂的亲缘关系。用Kinship可以求得一个加性遗传效应值/育种值,以此作为随机效应。考虑了群体结构和Kinship的模型就是混合线性模型MLM。
46.利用GCTA工具计算复杂性状/特征的遗传相关性
GCTA,是一款基于全基因组关联分析发展的分析工具,除了计算不同性状/表型间(traits)的遗传相关性外,还可以计算亲缘关系、近交系数。
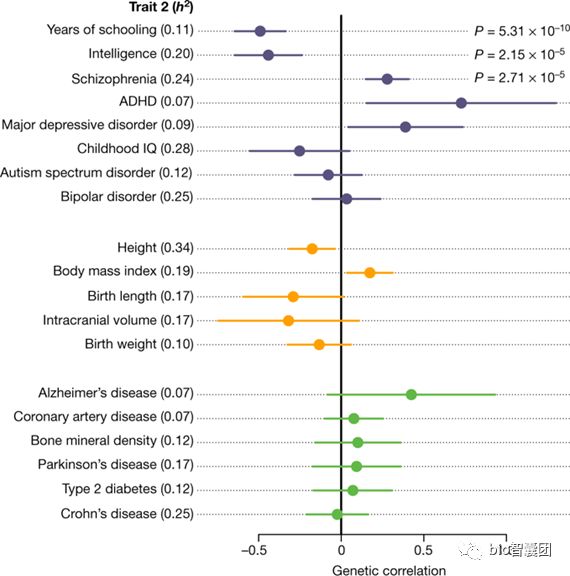
47.GWAS后续分析:LocusZoom图的绘制
LocusZoom图几乎是GWAS文章的必备图形之一,其主要作用是可以快速可视化GWAS找出来的信号在基因组的具体信息:比如周围有没有高度连锁的位点,高度连锁的位点是否也显著。
48.多基因风险评分(Polygenic Risk Score)的计算
传统的GWAS研究只计算单个SNP位点与表型之间的关联性,再用Bonferroni校正,通过给定的阈值,筛选出显著的SNP位点。
这样会存在两个问题,第一、Bonferroni校正非常严格,很多对表型有贡献的位点会因为达不到阈值而被过滤掉。第二、单个位点对表型的解释度是很低的,尤其是对于高血压这种多基因控制的表型,用一个个单独的位点解释高血压患病风险,就显得很单薄。
因此,开发一个能让我们直观的感受,患某种疾病的风险多高的工具,显然是非常有必要的。计算PRS的主流软件有PRSice,截止目前为止,该引用率有366次。
49.孟德尔随机化分析
50.用LDSC计算基因多效性、遗传度、遗传相关性
51.SNP功能注释网站合集
52.Fine-mapping相关的知识1
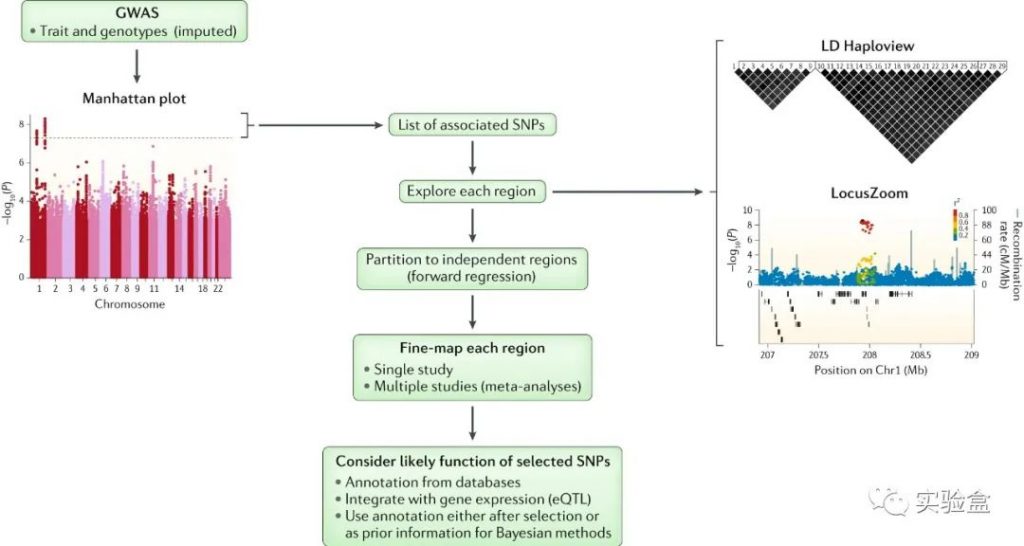
&emsp曼哈顿图中的点代表的不是样品,而是SNP。曼哈顿图中,显著的SNP并不是鹤立鸡群的冒出来,而是似乎被捧出来的,就像高楼大厦一样,从底下逐步冒出来的。这一座大厦其实就是连锁在一起的SNP,具有很高的LD score。虽然曼哈顿图里每个点是SNP,但是通常都会把最显著的SNP指向某个基因,因为大家最关注的还是SNP的致病根源,但这样找出来的只有编码区的SNP。最突出的SNP极有可能不是causal SNP,它只是near the causal SNP。问题就来了,怎么找causal SNP呢?fine mapping
&emsp一个常识就是GWAS是2007年才出现得,所以2017年才出了篇有名的综述ten years of GWAS,fine mapping是GWAS后才出现得。fine mapping目的很简单:GWAS找到的大多不是causal variants,fine mapping就是就fill这个gap。GWAS得到大体的SNP后,必须做两方面的深入分析:第一步就是对SNP给一个概率上的causality,这就是fine-mapping;第二步就是根据功能注释来确定该SNP确实能导致某个基因。
目前主要的的Statistical方法有两种,分别是:
- (1)对lead SNP的P值排序或LD分析;
- (2)基于贝叶斯方法计算posterior causality probabilities。
Fine-mapping详细介绍1、Fine-mapping详细介绍2、Fine-mapping详细介绍3、Fine-mapping详细介绍4、Fine-mapping详细介绍5
53.Fine-mapping相关的知识2
有了GWAS公共数据后的下一步就是找因果变异(causal varision),这篇文章介绍fine-mapping精细映射 和gene prioritization基因排序,简单一句话就是 translate GWAS loci to a functional understanding of the associated trait, while taking cell-type- and disease-specific context into account. 再简单点,搞清楚loci——trait的关系是不是因果。内容来自2020年1月Open Biology上发表的综述A practical view of fine-mapping and gene prioritization in the post-genome-wide association era。(注:涉及到因果就很麻烦,一对一,一对多,多对一,多对多,中介、调和、介导、反向等等等等,之后和孟德尔随机化、结构方程模型一起慢慢总结吧)。
表型性状phenotypic traits 和疾病都含有有影响其发育,易感性或特征的遗传成分。genetic regions (loci)基因座与表型性状相连接,GWAS通过比较和关联健康(baseline)人群和具有感兴趣性状特征(如1型糖尿病、乳糜泻和身高)的人群,获得数百万个相对常见的遗传变异(SNP)。通过GWAS获得的性状相关的遗传基因座,会被变异标记为一个marker或者top 变异。每个标记变异表示一个单体型,其中包含许多附近位于高度连锁不平衡(LD)变异,表明它们最有可能一起遗传。
Fine-mapping变异的方法共有五种:(1)找与功能元件的overlap;(2)利用等位基因特异性变异的效应;(3)找破坏TFBS转录因子结合位点的变异;(4)是直接测量变异对调控区域强度的影响;(5)利用3d交互找因果变异对基因表达的影响。
54.FINEMAP:使用GWAS摘要数据进行无功能注释数据的精细定位(Fine-mapping)
全基因组关联分析(GWAS)是非常流行的定位表型或疾病遗传位点方法。不过很多情况下,GWAS 发现的最显著的 SNP(top SNP 或者 index SNP)并不是真正造成影响的causal SNP(因果SNP),而是因为跟 causal SNP 之间存在的 LD 而变得显著。因而,后续还需要对结果进行 fine-mapping(精细定位),把 causal SNP找到。
如果想了解更多 fine-mapping 的知识,推荐看看 Nature Reviews Genetics From genome-wide associations to candidate causal variants by statistical fine-mapping 这篇综述。
55.CAUSALdb:涵盖数千个GWAS研究和Fine-mapping结果的可视化数据库
现在 GWAS 研究越来越多。要查询以往的 GWAS 研究结果,可以使用 GWAS Catalog。GWAS Catalog 包含的信息非常多,不过有时可能满足不了需要。这里,推荐一个叫 CAUSALdb 的数据库。
CAUSALdb(http://mulinlab.tmu.edu.cn/causaldb/index.html)是一个整合了大量人类 GWAS 摘要统计信息,并通过统一处理进行 fine-mapping 识别因果变异的数据库。
CAUSALdb 包含 3,000多个 GWAS 公共数据,并且该数量也在不断更新中。
它使用了PAINTOR、CAVIARBF 和 FINEMAP 三个最新的fine-mapping工具,估算 GWAS 候选位点中所有遗传变异的因果概率。这些结果,可以在交互式网页中查看。
56.单倍型(Haplotype)
基因在一条染色体上的组合称单倍型,在体细胞两条染色体上的组合称基因型,其表达的特异性称为表型。
更进一步的讲,单倍型也是指一个染色单体里面具有统计学关联性的一类单核苷酸多态性(SNPs)。一个单倍型内的这类统计学关联性和等位基因的确认被认为是可以明确的识别所有其他多态区域。这些信息对于探查普通疾病的基因学非常有用,也被用于人类单倍体型图计划(HapMap)中。
单倍型与SNP的区别
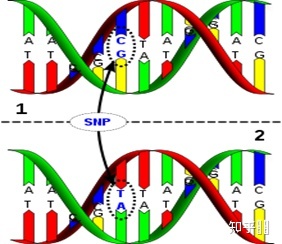
- SNP和单倍型的关系就是一个是一个碱基的突变,一个是一连串特定突变的位点的组合。
- SNP :是指基因组上由单个核苷酸的变异所引起的DNA序列多态性。如图:
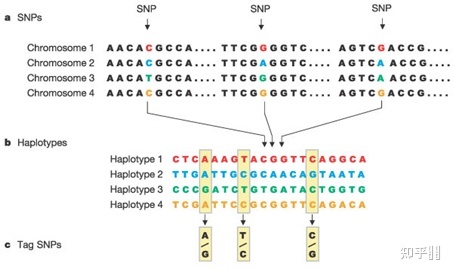
- 单倍型(haplotype):位于一条染色体上某一区域的一组相关联的多个SNP等位位点被称作单倍型。单倍型表示以基因 * 数字或数字字母组合表示,如CYP2C91,CYP2C9,如图:
57.GTEx、TCGA和ICGC数据库
(1)GTEx:Genotype-Tissue Expression
- GTEx收集的是正常人身上的组织来进行的测序,所以GTEx数据库包括的就只是正常人的数据。
- 一方面是可以研究正常人不同组织之间的基因表达的区别。
- 另一方面可以和TCGA联合使用。由于TCGA重点收集的还是癌症组织的数据,对于其正常的数据收集的相对来说较少,由于正常样本少所以对于差异表达的结果可能就不是很准确。这个时候如果我们把GTEx的数据纳入进来。这样分析的结果就会准确一些了。
(2)TCGA:The Cancer Genome Atlas(癌症基因组图谱)
- 肿瘤相关的数据库,包括了33个种肿瘤的数据。
- 数据库检测的数据类型多。对于同一个癌种,我们可以获得这个癌种的: 表达数据、miRNA表达数据、甲基化数据、突变数据和拷贝数数据。如果我们使用GEO数据库检索某一个癌种,同样也可以得到这些相关的数据。但是TCGA数据库珍贵的地方是,这个数据都是出自同一个人的。这样的话,我们就可以研究不同组学之间的交叉反应了。比如突变对于表达的影响、甲基化和表达的关系等等。
- TCGA除了包括了不同测序的数据,同时对于每一个纳入的患者还包括了其临床的信息。更难能可贵的是,临床信息当中还包括了预后随访的信息。这个我们就可以来分析以上的测序数据集和临床信息之间的关系了,比如分析基因表达和预后的关系等等。
(3)ICGC:International Cancer Genome Consortium(国际癌症基因组联盟)
- 这个数据库和TCGA的关系,就是ICGC数据库包括了TCGA的数据。另外呢,ICGC也纳入了其他别的地区所做的队列的测序数据。所以如果使用ICGC进行检索的话,我们可以得到更多的数据。
- ICGC是一个储存原始数据的地方,我们只需要检索相对应的关键词就可以得到具体的信息了。我们可以检索疾病、基因名称或者突变信息都可以。例如我们检索 gastric cancer,我们就可以得到这个联盟纳入的数据集。
58.eQTL(Expression quantitative trait loci)相关的文章
eQTL的全称是expression quantitative trait Loci,译为表达数量性状基因座,指的是染色体上一些能特定调控mRNA和蛋白质表达水平的区域,其mRNA/蛋白质的表达水平与数量性状成比例关系。其具体原理是把基因表达量作为一种数量性状,研究遗传突变与基因表达的相关性。
(1) eQTL的详细定义
表达数量性状基因座(eQTL)是对上述概念的进一步深化,它指的是染色体上一些能特定调控mRNA和蛋白质表达水平的区域,其mRNA/蛋白质的表达水平量与数量性状成比例关系。eQTL可分为顺式作用eQTL和反式作用eQTL,顺式作用eQTL就是某个基因的eQTL定位到该基因所在的基因组区域,表明可能是该基因本身的差别引起的mRNA水平变化;反式作用eQTL是指某个基因的eQTL定位到其他基因组区域,表明其他基因的差别控制该基因mRNA水平的差异。
其具体原理是把基因表达量作为一种数量性状,研究遗传突变与基因表达的相关性。

(2) QTL与eQTL的关系
数量性状基因座(QTL):控制数量性状的基因在基因组中的位置称数量性状基因座。常利用DNA分子标记技术对这些区域进行定位,与连续变化的数量性状表型有密切关系。
首先QTL是数量性状位点,比如身高是一个数量性状,其对应的控制基因的位点就是一个数量性状位点,而eQTL就是控制数量性状表达位点,即能控制数量性状基因(如身高基因)表达水平高低的那些基因的位点。
eQTL就是把基因表达作为一种性状,研究遗传突变与基因表达的相关性: 就好像研究遗传突变与身高的相关性一样。
早年可以通过同时做一个个体的SNP芯片和cDNA芯片, 在全基因组尺度研究突变与表达的相关性, 这种研究需要较多个体(例如1000个); 现在随着深度测序的出现,很多人开始用RNA-Seq在较少量个体中研究allele-specific expression,本质上就是eQTL。
简单地说, 遗传学研究经常发现一些致病或易感突变, 这些突变怎样导致表型有时候不太直观; 所以用某个基因的差异表达作为过渡: 突变A–>B基因表达变化–>表型;
(3) GTEx的前世今生
然而,事情却没有那么简单,从基因的改变到疾病等现象的出现,中间缺失了重要的一环,那就是基因的表达。也许在测序中,我们可以看到某一个基因上某一个位置的变化(比如说SNP单核苷酸变化),但是这种变化并不一定会影响mRNA的产生或者蛋白的改变。也就有可能不会影响到疾病或其他生物学过程。于是科学家想到了另一个指标——mRNA的序列数据。因为只有被表转译到mRNA上的基因,才可能进一步表达为蛋白(图1)。
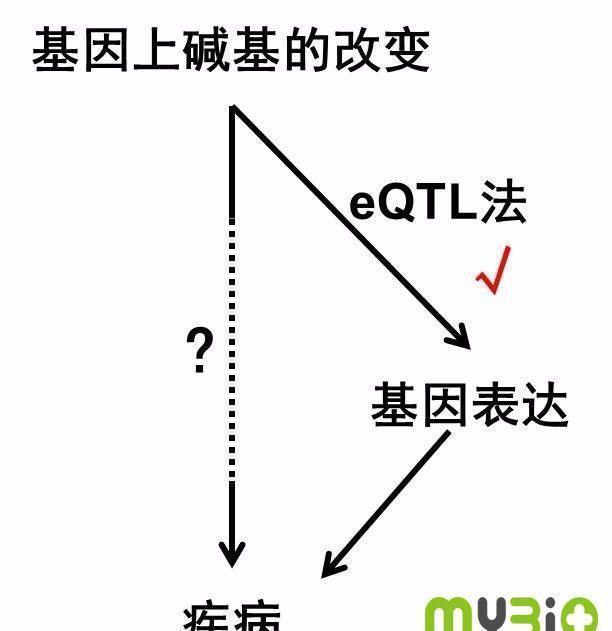
图1:eQTL是沟通基因改变与疾病的桥梁
但是要怎么搞清DNA改变是怎么影响mRNA的出现呢?这一过程被称为Expression quantitative trait loci(eQTL) 分析,目的在于得到单个DNA突变与单个基因表达量之间的相关性。与单个基因mRNA表达量相关的DNA突变,就被称为eQTL。
简单来讲,(并不简单,小编注)我们首先通过全基因组测序获得每个个体的DNA全序,然后以同种族的其他个体作为参照,标记出该个体所有的DNA变异位点, 称为SNP位点。同时,我们通过全基因组mRNA表达量测序得到该个体的特定组织样本中的基因表达量。以全部DNA变异位点为自变量,轮流以每种mRNA表达量为因变量,用大量的个体数据做样本进行线性回归,就可以得到每一个SNP位点和每一个mRNA表达量之间的关系。
GTEx是第一个收集了多个人体器官mRNA测序的数据库,并提供了跨器官的eQTL研究平台。
当前使用的GTEx v6p版本的原始数据来自于449名生前健康的遗体捐献者的44个不同的器官。图2是不同器官里面样本数的直观展示。由这个图可以看出,这一数据库中涉及的数据覆盖面非常广,数据量大,具有重要的应用潜力。
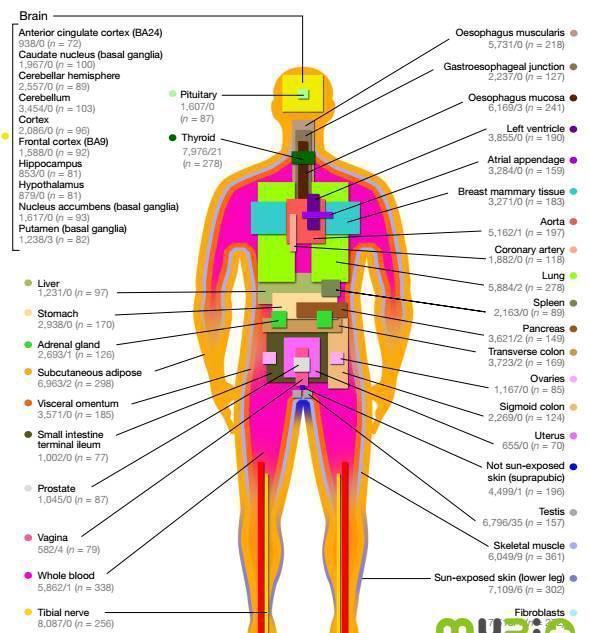
图2:GTEx 样品取材来源图示。灰色字体为 cis-eQTL 数/trans-eQTL数
GTEx 为挖掘器官特异的基因组数据提供了一个非常好的平台。这是目前唯一一个可以提供这些内容的数据库工具。有了这些,科学家就可以尝试回答很多问题:比如基因相互作用的网络在不同器官里会有怎么样不同的表现?不同组织中基因突变对于基因表达有哪些影响?特定染色体在不同的器官中作用有哪些?等等。
更详细的内容请见上述的链接。
59.Plink格式详解
- ped 和 map 是一组的
- bed fam bim 是一组的
60.知道了SNP,如何查看candidate基因以及对应的氨基酸突变等信息?
If there are several significant hits in a region, select the one with lowest p-value. Copy each SNP name and search on genetic databases such as dbSNP, UCSC genome browser, HaploReg2. Is it on a gene? Is it exonic? Is it non-synonymous? Is it functional?
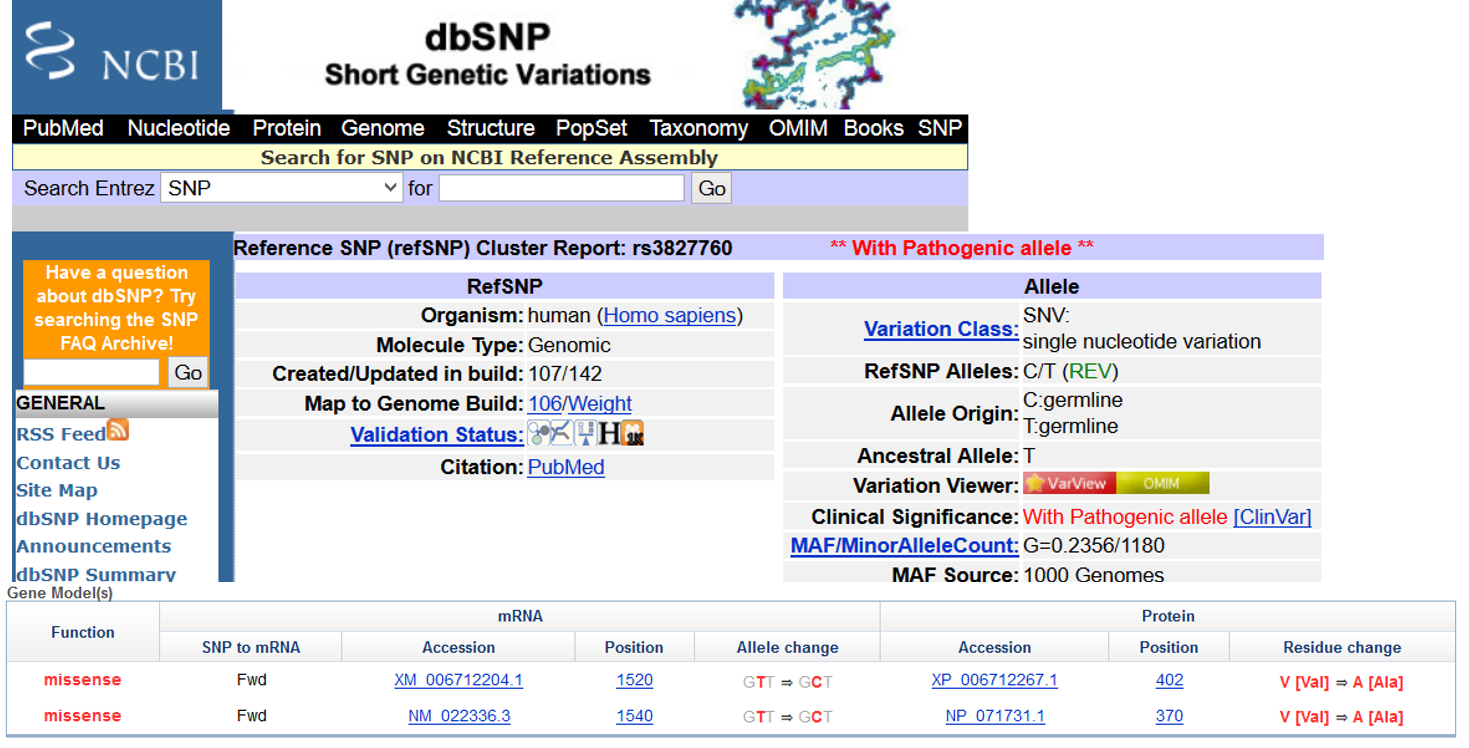
Also check allele frequency differences in continental populations. Large differences means that this SNP has diversity across populations.