1.Title: “Pain, analgesia and genetics (2011).”
1.1 Abstract
Pioneering work undertaken in mice more than a decade ago, showed a strong genetic contribution to levels of nociception/hypersensitivity as well as levels of antinociception produced by commonly available analgesic agents. To date more than 300 candidate ‘pain’ genes have been identified as potentially contributing to heritable differences in pain sensitivity and analgesic responsiveness in animals and humans, with this information available in a publicly accessible database. Since then, many genetic association studies have been conducted in humans to investigate the possibility that single nucleotide polymorphisms (SNPs) in an individual gene may explain drug inefficacy or excessive toxicity experienced by a small subset of the whole population who have the rare allele for a particular SNP.
1.2 Summary
Despite the fact that SNPs in more than 20 genes that affect pain sensitivity or contribute to interindividual variability in responses to analgesic medications have been identified in the human genome, much of the data is conflicting.
1.3 Introduction
Globally, the prevalence of chronic pain is high at 15–20% of the adult population, with pain severity ratings given by patients encompassing not only the intensity of the nociceptive stimulus but also the individual’s affective/emotional response to that stimulus. This in turn results in marked interindividual variability in reported levels of pain intensity for apparently similar pain states.
1.4 Rodent Studies and Pain Genetics
Pioneering work by Mogil et al. just over a decade ago using quantitative sensory trait analysis in 11 different mouse strains across 12 different testing modalities, showed clear heritability of pain-related traits when assessed using thermal, mechanical and chemical measures of nociception. Impressively, they found a 30–76% genetic contribution to levels of nociception/hypersensitivity in their mouse studies.
1.5 Single Nucleotide Polymorphisms in Target Genes
In humans, a single nucleotide polymorphism (SNP) is a DNA sequence variation at a specific location in the genome, occurring when a single nucleotide (A, T, C or G) differs between individuals at a frequency of more than 1% in the normal population, such that for each SNP there will be two possible alleles. The possibility that individual SNPs may explain lack of efficacy or excessive toxicity of a medication in those individuals with the rare allele for a particular SNP, has given rise to the field of pharmacogenetics. Although SNPs have been identified in more than 20 genes that affect pain sensitivity and/or contribute to interindividual variability in responses to analgesic medications, much of the data are conflicting.
1.6 Genetics and Pain in Humans
- Pain insensitivity: loss of function mutations
These rare pain phenotypes involve loss-of-function mutations in individual genes including SCN9A, SPTLC1,WNK1/HSN2,IKBKAP,NTRK1 and NGFB that encode ion channels, enzymes, transcription factors and neurotrophins.
- Pain sensitivity: gain of function mutations
Gain-of-function mutations in the Nav1.7 gene (SCN9A) that increase the excitability of dorsal root ganglia neurons are linked to two clinically-distinct familial pain syndromes (Table 1), viz inherited erythromelalgia (IEM) and paroxys-mal extreme pain disorder (PEPD).
1.7 Quantitative sensory testing: volunteer studies
In volunteer studies, quantitative sensory testing (QST) is used to determine experimental pain thresholds or stimulus response curves for sensory processing across a range of pain modalities, including thermal, mechanical, electrical and chemical stimuli. More sophisticated designs employing QST involve application of these stimuli to a range of tissue types, including skin, muscles and viscera to produce a mosaic of responses. In recent years, QST has been used in volunteer studies to assess the contribution of the genetic component to interindividual variability in pain thresholds.
1.8 Does pre-operative quantitative sensory testinghave clinical relevance?
Pre-operative QST responses were able to predict 4–54% of the variance in the postoperative clinical pain experience depending upon the stimulation methods and the test paradigm utilized.
- Human twin studies
In a QST study undertaken in 98 pairs of volunteer healthy female twins (51 monozygotic and 47 dizygotic) using a wide range of noxious stimuli, genetic components were able to explain 22–55% of interindividual variability for the majority of painful stimuli assessed, particularly heat and chemical pain thresholds. More recently, in a study involving 53 pairs of monozygotic and 39 pairs of dizygotic twins, QST revealed similar findings in that 60% of the variance in cold pressor pain responses and 26% of the variance in heat pain responses could be explained by genetic factors. Clearly, as cold pressor pain and contact heat pain are distinct phenomena from both the genetic and environmental perspectives, this cautions against generalizing genetic findings from one pain modality to another.
1.9 Genetic association studies in humans
Genes investigated in thisway include CYP2D6, CYP3A4, CYP2C9, CYP2C19 and UGT2B7 that encode key enzymes in drug metabolism, ABCB1 that encodes the P-glycoprotein transporter, HTT that encodes the serotonin transporter, SLC6A2 that encodes the noradrenaline transporter, as well those encoding the a-subunit of the Nav1.7 sodium channel (SCN9A), them-opioid receptor (OPRM1), the melanocortin-1 receptor(MC1R), the b2-adrenergic receptor (ADRB2), cytokines (IL-1A/B, IL-6, TNFA, ILIRN, IL10) and the enzymes catechol-O-methyltransferase (COMT) and guanosine triphosphate cyclohydrolase 1 (GCH1), to name but a few.
1.10 Genetic Association Studies of Pain Sensitivity and Analgesic Drug Pharmacodynamics
- Melanocortin-1 receptor (MC1R)
- Guanosine triphosphate cyclohydrolase 1 (GCH1)
- SCN9A (Nav1.7 sodium channel)
- OPRM1 (µ-opioid receptor)
- Serotonin receptor
- Catecholamine neurotransmitters and pain: SERT, NET, COMT, MAO
1.11 Genetic association studies and analgesic drug pharmacokinetics
- Cytochrome P450 enzymes
- CYP2D6 (cytochrome P450 2D6)
- CPY3A (cytochrome P450 3A)
- CYP3A4 (cytochrome P450 3A4) and CYP3A5 (cytochrome P450 3A5)
- CYP2C9 (cytochrome P450 2C9)
- CYP2C19 (cytochrome P450 2C19)
- Phase 2 drug metabolizing enzymes: the uridine diphosphoglucuronosyl transferase superfamily
- Drug transporters in the blood–brain barrier
1.12 Genetic association studies and pain – overview
Despite initially promising results, most genetic association studies performed over the past decade to assess genetic contributions to pain phenotypes in patients, have either failed to replicate or have been only partially replicated. Possible reasons include considerable between-study variability in their design (underpowered studies), execution problems (heterogenous study populations, poor phenotyping, genotyping errors), unsuitable choice of statistical methods (failure to correct for multiple comparisons), as well as between-study differences in interacting environmental factors that affect pain phenotypes.
1.13 Genome-wide association studies: insights from other fields
Over the past two years, a number of genome-wide association studies (GWAS) for complex diseases such as diabetes have been undertaken, resulting in more than 250 genetic loci in which common genetic variants appear to be reproducibly associated with polygenic traits. However, the effect sizes for common variants, both individually and in combination, are modest, likely contributing less than 1% of phenotypic variation. Even with highly heritable phenotypes such as height (heritability estimates at~90%), the most significant SNP from a GWAS of ~5000 individuals followed by extensive genotyping with~20 000 people could only explain 0.3% of the interindividual variation. Hence, by extrapolation, it is likely that a GWAS in the pain field would produce broadly similar results, with a large number of genes each contributing a small amount to interindividual variability in pain sensitivity and analgesic dosing requirements.
1.14 Gene environment communication: epigenetics and RNA-editing
In the pain genetics field to date, the primary focus has been on assessing the impact of SNPs on protein-coding regions of the genome, which in total account for only 2% of the mammalian genome. However, many SNPs also occur in non-protein coding regions and recent research from other fields sheds light on this emerging dimension of pain genetics.
1.15 Epigenetics
In nerve-injured mice, there was epigenetic silencing of OPRM1, SCN10A and KCND3 genes in dorsal root ganglia to produce long-lasting downregulation of expression levels of the corresponding protein products, viz m-opioid receptors, Nav1.8 sodium channels and Kv4.3 potassium channels, respectively, all of which have key roles in pain modulation.
1.16 Conclusions
Interindividual variability in pain sensitivity and analgesic drug responsiveness in the clinical setting appears to be underpinned by complex interactions between an array of genetic and environmental factors. Recent research from other fields implicates epigenetic mechanisms including RNA editing as a means for facilitating dynamic gene-environment communication.
Be underpinned by:以。。。为基础
2.Title:” Pain Genetics basic to translational science (2014).”
2.1 How Do Pain Genes Affect Pain Experience
- (1)Variability in Pain Experience
There is remarkable variability in the amount pain that different people experience. Variability is the rule for acute pain in response to noxious stimuli; we each have our own “pain threshold.”
When trying to account for the variability of pain, there is no denying the importance of environmental factors. But environment isn’t everything. Pain genetics is premised on the idea that genetic factors contribute as much to individual differences in pain response as the environment, for some traits perhaps even more.
- (2) Heritability of Pain: Historical Roots
Most biological traits are affected by genes, environment, and interactions between the two. It should therefore come as no surprise that pain response is likewise affected by nature as well as nurture. Nonetheless, because of the striking influence of socialization in our day-to-day pain experience, the detection of an important genetic component took many people by surprise. The first solid indications of heritability came from rare familial disorders such as congenital insensitivity to pain with anhidrosis (CIPA), in which affected family members feel no pain, and Fabry’s disease and familial hemiplegic migraine (FHM) where affected family members suffer from extreme pain. The simple pattern of inheritance typical of such conditions points to the Mendelian inheritance of a single mutant gene of large effect. Using “linkage analysis,” today it is fairly straightforward to identify the specific causative mutant gene and the particular relevant base-pair change(s) within the gene that cause the disease. For Mendelian traits, the causative gene has already been pinpointed for all but the most obscure pain conditions.
Evidence for a genetic predisposition to pain on noxious stimulation in healthy individuals, and in painful medical conditions in which inheritance is non-Mendelian, came much later. Historically, the most common source of information on these (presumably polygenic) pain traits came from epidemiological research and the study of twins. For example, there are reports from the early 1900s of families with a higher-than-expected incidence of TN. Likewise, it was noticed that pain as a normal trait (e.g., in response to experimentally applied heat and cold in healthy individuals) and pain in common medical conditions such as backache and sciatica are more concordant in identical (monozygotic) than in fraternal (dizygotic).
Unlike Mendelian pain traits, it is still not straightforward to identify the genes responsible for these more complex traits. This is because they are caused by a combination of gene polymorphisms whose individual effects may be small. In addition, the pain traits themselves are not black and white. Pain is typically graded in intensity, and often inheritance is manifest as a statistical predisposition to developing a painful condition under particular circumstances. In recent years, the “association study” has emerged as an approach to identifying the genes involved in non-Mendelian inheritance. In association studies, cohorts of unrelated individuals with contrasting pain phenotype are compared for genetic differences. Ideally, all of the genes in the genome are searched for consistent differences between the cohort with and the cohort without pain (genome-wide association study (GWAS)). However, because of the high cost of carrying out a GWAS, comparisons are usually limited to one or more gene “candidates” that the investigator has prior reason to think might be relevant. A hybrid between these two methods is noted briefly in the succeeding text, where GWAS analysis was carried out inexpensively in mice using a neuropathic pain model, and the pain gene discovered was then tested as a candidate in a human association study.
- (3) Why is Pain Genetics Interesting and Potentially Useful?
There are basically four reasons why a pain professional ought to be interested to know that pain response is affected by genes:
- 1) Stigma:
- 2) Diagnosis, prognosis, and guidance:
Polymorphisms (variants) in pain genes affect pain response. Typically these polymorphisms amount to single-letter differences in the nucleotide sequence of A’s, T’s, G’s, and C’s that make up the genome. Such single base-pair differences are called single-nucleotide polymorphisms (SNPs). Quite a few SNPs have already been identified that might affect the amount of pain that an individual suffers. Although we are not there yet, it is fairly straightforward to identify, in a simple blood or saliva test, whether an individual patient carries one or more pain-related SNPs that might account for exacerbated pain. It can be anticipated that in the future, results of such lab tests will assist in the accurate medical diagnosis of the underlying pain condition and in making a prognosis. Such information may also guide treatment by genetic counseling or even form the basis of gene therapy.
It is a gene called Cacng2 and is known to code for the gamma subunit of voltage-sensitive Ca2+ channels (the stargazin protein). Cacng2 had previously been implicated in epileptogen-esis, but not in neuropathic pain. Virtually all mouse genes have a homolog in the human genome, and Cacng2 is no exception. The human version of the Cacng2 gene is designated CACNG2.
- 3) Pharmacogenetics and individualized medicine:
A third arena in which pain genetics could make a difference is in predicting which patients are likely to respond to which analgesic drugs.
-
4) Discovery of novel pain mechanisms
-
(4) What Are Pain Genes?
Genes are parts of DNA molecules that contain the information used by cells to construct protein molecules. They “code” for proteins. Proteins are “products” of gene transcription and transla-tion, the two cellular processes that exploit the information encoded in the sequence of nucleotide base pairs in the DNA molecule to manufacture particular proteins. It is these proteins that carry out the work of the cell, such as enzymatic action, motility, and electrical impulse generation. It is essential to realize that genes code for protein molecules, not for sensory and emotional experiences such as pain. Pain and these other traits are complex downstream effects of protein function. Variation in the action of certain protein species makes a difference to how pain is perceived by a conscious brain, while variation of other proteins doesn’t. A “pain gene” is a gene for which there are one or more polymorphisms (i.e., variations in the sequence of DNA base pairs) that affect the expression or the functioning of its protein product in a way that affects pain response.
- (5) How Many Pain Genes Are There?
DNA sequence variants (SNPs) that are common among humans are called “polymorphisms.” Rare variants, occurring in <1% of the population, are called “mutations.” Genes that differ slightly because of these variants are “alleles” of the gene. Nonetheless, only a tiny fraction of all genes have allelic variations with known effects on either cellular function, organ function, or the behavior of the individual, including pain response. Although more will surely be found, it is a safe bet that overall, most allelic variants in humans will prove to have no significant effects on pain processing. Only a small fraction of human genes are likely to be pain genes. However, some allelic variants in some genes do have measurable effects on pain, and the effect is sometimes substantial. As noted earlier, individual SNPs cause certain Mendelian diseases associated with severe pain or congenital insensitivity to pain. A particularly interesting example involves the SCN9A gene that codes for the alpha-subunit protein of the voltage-gated Na+ channel Nav1.7. Some alleles of this gene cause severe pain, others cause complete pain insensitivity, and still others only slightly affect the likelihood of an individual’s developing one or another painful condition. Evolution has probably weeded out from the genome most alleles that directly cause painful diseases, but many alleles remain that can predispose to conditions with a pain signature.
Overall, putting aside the rare Mendelian mutations, pain phenotype is likely determined by polymorphisms in a few hundred genes, with only a fraction of these contributing in any particular individual or painful condition. Some pain genes enhance pain (or the likelihood of developing pain), and some protect from it. Some likely have a relatively large effect, and some have a small effect. Complicating things still further, it is unlikely that the final pain phenotype results from simple summation of the various allelic effects present. There is probably a complex calculus where the various alleles inherited by an individual interact with one another nonlinearly and with the environment. Thus, for example, a genetic polymorphism may have no effect on pain response except under unusual environmental circumstances (e.g., at high altitudes), or if your kids made you particularly exasperated. After all, pain genes are of medical and scientific interest to the extent that individual allelic variants have a reasonably strong and predictive effect on pain response. It is not enough to find pain genes. One wants to find significant ones. This will require additional work. It is possible to incorporate in the search for pain genes strategies for enhancing the likelihood of finding functionally significant polymorphisms. One such strategy is the mouse–human hybrid approach noted earlier. It is much easier to screen mice for major heritable pain phenotypes than to screen people.
- (6) How Do Pain Genes Affect Pain Experience?
Genetic polymorphisms do not affect pain response by an action on the psyche, but rather by altering cellular functions. They can do this in several ways. Some polymorphisms occur in the nucleotide sequence that actually encodes for the gene’s protein product (i.e., in exons). Depending on the details, this may alter the amino acid sequence of the protein, changing its shape or charge configu-ration and hence its functioning. A polymorphism of a single nucleotide (SNP) in the exonic sequence can be enough to significantly alter a protein’s function, depressing it, enhancing it, or eliminating it entirely. Exonic polymorphisms can also affect the “bar code” address of a protein, affecting “trafficking.” This is the process that determines where in the neuron the protein will be delivered. The affected protein may work normally, but if it is sent to the wrong part of the cell, it can’t do its job properly. Sequence polymorphisms that affect pain can also occur in noncoding parts of a gene (introns) and noncoding intergenic regions. Such polymorphisms can affect the protein product itself by altering gene splicing. But more commonly the effect of non-exonic polymorphisms is on the regulation of gene expression, for example, changing how much of the gene product is synthesized (copy number, abundance).
- (7) Direct Effects of Allelic Variation on Pain Mechanisms
By altering cellular functions, pain genes can affect pain response in a direct manner or indirectly. Typical direct mechanisms include (i) increased intrinsic excitability of pain signaling neurons, due, for example, to altered ion channels and membrane receptors, or (ii) hyperexcitability in pain processing networks due, for example, to reduced inhibitory neurotransmission. Neuronal and network excitability are determined by a very large number of different molecules. In principle, genetic polymorphisms affecting any of them could directly affect pain. It is likely that only a small fraction of the genes that build the pain system actually contribute to individual pain variability. The Pain Genes Database currently lists 390 genes that yield a pain phenotype in transgenic mice. Many of these genetic variants probably do not occur in the homologous gene in human populations.
Sometimes, however, a functional allelic variant occurs in a gene that sits at a nodal point in pain physiology. A prime example is the Nav1.7 Na+ channel gene (SCN9A). As noted earlier, some mutations in this gene cause the extreme pain conditions familial erythromelalgia and paroxysmal extreme pain disorder. Other mutations in the same gene produce congenital insensitivity to pain. Investigators have expressed both the pain-inducing and the pain-preventing mutations of the human SCN9A gene in rodent sensory neurons and in human nonneural cells that do not otherwise express Na+ channels. Recordings from these cells showed that the pain-inducing mutations render the neurons electrically hyperexcitable, prone to generate nerve impulses in excess. They are “gain-of-function” mutations. In contrast, the mutations that produced pain insensitivity in humans caused experimentally modified cells to be hypo-excitable (“loss-of-function” mutations). What is more, the cellular hyperexcitability and hypo-excitability have been directly linked to relevant alterations in specific parts of the Na+ channel molecule itself. This is a rare example where clinical pain phenotypes can be directly associated with point mutations in a particu-lar gene and its protein product. It is also a highly informative example, adding considerably to our understanding of Na+ channel gating and pain processing.
Polymorphisms also occur in the genes that code for types of Na+ channels other than Nav1.7. These, however, apparently do not cause severe pain or pain insensitivity; at least no such families have been identified yet. Surveying common polymorphisms of small effect in genes that have already been found to carry rare mutations of large effect may be a productive strategy in general.
As strong as the case is for SCN9A variants having a direct action on pain, the information in hand nonetheless leaves major unknowns. For example, the nature of the mutations does not explain why in erythromelalgia pain occurs mainly in the hands and feet (the mutation affects Nav1.7 proteins everywhere), why different gain-of-function mutations in the same gene cause very different clinical symptoms (erythromelalgia vs. paroxysmal extreme pain disorder), or why the loss of Nav1.7 function leads to pain insensitivity, but does not cause a deficit in nerve conduction or sensation in most other modalities (touch, vibration, vision).
- (8) Indirect Effects of Allelic Variation on Pain Mechanisms
Allelic variants sometimes have effects that appear on the face of it to link directly to pain mechanisms, but in fact don’t. Consider an oncogene which causes the formation of a tumor that compresses a nerve and induces a painful compression neuropathy. One of the major potential contributions of pain genetics is to highlight genes that are empirically (clinically) important for pain experience. The challenge for pain scientists is then to determine how these genes cause the effects that they cause.
Most allelic variations that tip the balance toward pain can be expected to operate in ways that are indirect.
- (9) Disease Susceptibility Genes Versus Pain Susceptibility Genes
Identification of disease susceptibility genes may be of considerable medical interest for disease diagnosis and prevention, but such genes are not certain to contribute much to an understanding of the underlying pain process. It is difficult to know in advance. Therefore, if the aim is to understand pain rather than disease, a more likely approach is to identify genes in which sequence variants cause one person to develop pain, while another doesn’t, in the presence of the same disease or injury. These are “pain susceptibility genes.”
Finding genes that alter pain response to experimentally applied stimuli is relatively straight-forward using the GWAS approach.
- (10) Perspective
One reason is that the methods and the choice of comparison groups used to date have often not been optimal. This includes both the underpowered scope of many of the trials undertaken so far and the focus on candidate genes selected on the basis of preconceptions rather than unbiased GWAS. In particular, one needs to recall that genes which predispose to developing a disease that tends to be painful may be only distantly related to the neural mechanisms of pain processing itself. This makes for weak genetic inference.
2.2 Conservation of Pain Genes Across Evolution
- (1) Introduction
The ability of an organism to sense and respond to noxious temperature, mechanical insult, and chemical hazards is crucial for the survival of all animals. These basic nociceptive behaviors are not unique to higher organisms but also exist in lower organisms. Interestingly, besides the similarity in basic nociceptive behaviors seen in many animal species, recent studies have shown that essential genetic components associated with multiple nociception processes are conserved.
- (2) Anatomical Organization of Nociception Apparatus in Mammals and Drosophila
In mammals, detection and transduction of painful stimuli rely on free nerve endings (nociceptors) that consist of classes of myelinated or unmyelinated fibers. The first class consists of the myelinated fibers Aδ and Aβ. Aδ-fibers are characterized by thin axons and myelin sheaths and high conduction velocity, and these fibers are responsible for “fast pain” that is well localized. In contrast to Aδ-fibers, Aβ-fibers have a large diameter and often respond to innocuous stimulation but seem to transduce painful signals following nerve injury. The second class of nociceptors consists of small-diameter C-fibers that are unmyelinated, have slow conduction velocity, and transmit “slow pain” that is poorly localized.
- (3) Acute Heat Pain in Mammals
In mammals, acute thermal pain sensation is mediated by several transient receptor potential (TRP) channels. Among those are TRP vanilloid-1 (TRPV1) and TRP vanilloid-2 (TRPV2) which are found to respond to temperature above 43 °C and 52 °C, respectively. TRPV1 is expressed in both Aδ- and C-fibers, and its expression is increased in response to heat-hypersensitivity-inducing agents such as neurotrophin and bradykinin, while TRPV2 is more wildly expressed and does not respond to capsaicin. Other TRPV channel subtypes including TRPV3 and TRPV4 are also heat-gated ion channels with thermal gating ranging from 25 °C to 35 °C. In addition to TRPV family members, TRP melastatin-3 (TRPM3) is also a thermosensitive channel, and TRPM3 knockout mice show profound deficits in their avoidance responses to noxious heat. Interestingly, temperature sensitivity profiles of various TRP channels appear to be flexible across species, for example, TRPV3 activation threshold is 34 °C in mice but 16 °C in the western frog, whereas TRPV4 activation threshold was found to be 25 °C–33 °C in HEK293 cells but around 27 °C in Xenopus oocytes.
While significant progress has been made in understanding how TRP channels contribute to heat pain perception, recent molecular evidence coupled with functional MRI studies in transgenic mice has also highlighted a role for cerebral activity in regulating heat pain perception in mammals.
- (4) Acute Heat Nociception in Drosophila
Thermal nociception in Drosophila is regulated by several members of the TRP channel family including TRP ankyrin-1 (TRPA1), painless, and pyrexia. In addition to these TRP channel family genes, stj (α2δ3), which encodes a peripheral subunit of multiple Ca2+ channels and important for mouse and human heat pain, also plays an important role in acute heat nociception in flies. Another gene, amnesiac (amn), which encodes a putative neuropeptide precursor known to be involved in learning and memory, has also been implicated in fly heat nociception, although the mechanism of action in nociception has yet to be explored.
Though painless and amn are not evolutionarily conserved, TRPA1 and stj have likely mammalian orthologs. This suggests the possibility that core genetic components of thermal perception are conserved through species; however, genetic variation in these channels may lead to variations in their thermal response profiles across phyla.
- (5) Mechanical Pain in Mammals
Mechanical stimuli cause the opening of mechanosensitive ion channels at sensory nerve endings leading to an action potential that transduces the sense of touch and mechanical pain to the CNS. Noxious mechanically evoked action potentials are transmitted by myelinated Aδ-fiber and unmyelinated C-fiber neurons in mammals. Though several candidate genes have been proposed to be mechanical pain mediators, genetic components involved in mechanical pain in mammals remain vague.
- (6) Mechanical Nociception in Drosophila
One fly mechanical nociception gene is pickpocket (ppk), which encodes a degenerin/epithelial sodium channel (DEG/ENaC) subunit. Painless, which is expressed in all md neurons, is also required for sensing noxious mechanical input. Another Drosophila mechanotransducer is no mechanoreceptor potential C (NOMPC), a member of TRP, which is expressed in class I dendritic arborization neuron and is responsible for sensing light touch. The heat pain gene TRPA1 also appears to play a conserved role in mechanical nociception, and similar to some reports for TRPA1 knockout mice.
- (7) Chemical Nociception in Mammals
The ability to detect noxious chemicals is crucial and also elicits pain and nociceptor sensitization, since these substances can cause tissue damage especially of mucous membranes, for example, in the eyes, mouth, and respiratory tract. In mammals, TRP channels function as cognate receptors for a number of plant-derived products including menthol in mint, allicin in garlic, hydroxy-a-sanshool in prickly ash, allyl isothiocyanate (AITC) in wasabi, and mustard oil. Among these channels, TRPA1 has emerged as an important player in chemical nociception, and TRPA1 is activated in response to a variety of thiol-reactive compounds and environmental toxicants including volatile irritants such as acrolein (found in cigarette smoke), and TRPA1 is essential for acrolein’s actions on sensory neurons in mice. Mechanistically, these reactive substances can activate TRPA1 by forming covalent bonds with cysteine and lysine residues in the channel, inducing conformation changes in the channel’s N-terminal.
- (8) Chemical Nociception in Drosophila
In flies, chemical nociception is also mediated by TRPA1, and TRPA1 mutant flies fail to detect noxious chemical stimuli.
- (9) Inflammatory Pain in Mammals
Significant tissue damage often causes inflammation, which promotes healing and repair of the damaged tissue. However, tissue damage also often involves nociceptor sensitization, where sensory nerves undergo significant increases in responsiveness resulting in allodynia (pain due to a normally innocuous stimulus) and hyperalgesia (heightened pain sensitivity to a noxious stimulus). This sensitization results from the increased expression of cell surface receptors, such as Mas-related G protein-coupled receptors (Mrgprs) and TRP channels, in response to accumulation of substances released from nociceptors and nonneural cells near the injured areas. These substances include substance P, bradykinin, prostaglandins, neurotrophins, cytokines, and chemokines, among others.
- (10) Persistent Pain in Drosophila
Consistent with the analogous inflammatory sensitization in mammalian systems, UV-induced sensitization was found to be modulated by Eiger (TNF family cytokine) released from damaged epidermal cells and signaling through Wengen (TNFR ortholog). These data suggest that a common mechanism of cytokine-mediated nociceptive sensitization is conserved across species.
- (11) Neuropathic Pain in Mammals
Neuropathic pain models in mammalian systems use manifestations of allodynia and hyperalgesia to thermal, cold, and mechanical stimuli as readouts of neuropathic pain. Common neuropathic pain models are chronic constriction injury (CCI) of the sciatic nerve and spared nerve injury (SNI) model.
- (12) Structural Reorganizations of Nerve Fibers in Neuropathic Pain
Following nerve injury, structural reorganizations at the cellular, molecular, and synaptic levels have been reported. Following injury, there is a neuronal regeneration process that involves reestablishment of axonal contact with the target and remyelination of regenerated axons. Activities of a range of trophic factors, such as IL-1β, TNF, NGF, and BDNF, known to regulate peripheral nerve regeneration after nerve injury dare upregulated by following injury. However, this regeneration does not always result in correct synaptic connections, and inappropriate reconnections formed during repair may be one mechanism causing neuropathic pain.
- (13) Mammalian Neuropathic Pain Genes That Are Conserved in Drosophila
In vivo pharmacological or transgenic knockout approaches have established that in mammals, neuropathic pain is regulated by several types of factors. These include members of the NGF/TNF family of cytokines and their receptors, a number of ion channels, and some additional factors.
- (14) Long-Term Potentiation and Long-Term Depression in Neuropathic Pain in Mammals
Long-term potentiation (LTP) describes a long-lasting increase in synaptic strength and is a candidate molecular mechanism to explain neuronal memory formation. LTP can be induced experimentally through high-frequency electrical stimulation or physiologically through stimuli that cause enhancement of neurotransmitter release in presynaptic sites or through postsynaptic re-enforcement. A growing body of research has suggested that LTP is also induced by tissue damage and acts as a mechanism underlying central sensitization in chronic pain.
In contrast to LTP, long-term depression (LTD) is defined as the long-lasting reduction in synaptic strength, and in C-fibers, LTD can be induced by low-frequency (1 Hz, 15 min) conditioning stimulation.
Induction of LTP and LTD occurs through the N-methyl-d-aspartate (NMDA)-type glutamate receptors, and targeting NMDARs has been suggested as an approach to manage central sensitization.
- (15) Neuropathic Pain in Drosophila
In Drosophila, a neuropathic pain model has not yet been developed; however, the neuronal processes associated with neuropathic pain have all been described in the fly.
- (16)Conclusions
Although the nervous system of invertebrate is relatively simple in structure, it shares with vertebrate common genetic mechanisms regulating acute and persistent pain perception. In addition, important cellular and molecular features suggested to regulate neuropathic pain in vertebrates are also found in Drosophila. Although the specific mechanisms at play can be different, many Drosophila mutations leading to abnormal nociceptive responses have orthologs that are also implicated in mammalian nociceptive disorders.
2.3 Defining Human Pain Phenotypes for Genetic Association Studies
- (1) Introduction
Pain can arise from any number of causes, in a variety of tissue types, is modulated at multiple levels of the nervous system, and is an aggregate experience composed of sensory, affective, and cognitive components, to mention some of the complexities. Added to this, pain researchers have developed a number of scales for measuring pain and a number of experimental models and other techniques for assessing the functioning of the pain system in part or as a whole. Phenotype selection is crucial to the success of genetic studies, and a poorly defined phenotype may result in study failure through nonsignificant findings or, possibly worse, significant but incomprehensible findings.
- (2) What is a Pain Phenotype?
Under normal circumstances pain occurs as a response to tissue damage. This response starts with the transduction of a neural signal at the primary afferent, which undergoes modulation at multiple levels of the nervous system until it ultimately results in the experience of pain and its expression. Though the tissue damage and pain expression are the immediately observable features of this process, they are, strictly speaking, not part of the pain system. Rather, the primary goal of pain research in general and of pain genetics in particular is to understand the processing that lies between these endpoints. Thus, a pain phenotype may be defined as a measure that directly or indirectly reflects the processing of parts or the whole of the pain system, excluding tissue pathology and pain expression.
A very important characteristic of the pain system is that its efficacy varies enormously across subjects. A noxious stimulus that some find unbearably painful will by others not be considered painful at all. We term these differences individual differences in pain sensitivity. The existence of large individual differences in pain sensitivity holds great promise for pain genetics. By identifying the molecular mechanisms that contribute to these differences, one may hope to develop analgesic drugs that target these molecules. Conversely, if individual differences in pain were non-existent, then pain genetics would be meaningless. Thus, a very important and necessary characteristic of a pain phenotype is its ability to show variation between subjects.
Pain sensitivity has normally been measured in experimental pain models, which have the great advantage of complete control over the noxious stimulus. However, the concept is equally applicable to clinical pain. For instance, among subjects suffering from the same clinical condition, the between-subjects variation in pain ratings frequently covers the entire pain scale, and for conditions where objective pathology is quantifiable, there is typically poor correlation between disease severity and pain magnitude. There is also substantial evidence that pain sensitivity as assessed with experimental measures is related to clinical pain. Most, if not all, chronic pain conditions are associated with increased pain sensitivity, and it is likely that sensitization of the pain system contributes to the degree of clinical pain. For neuropathic pain conditions, local alterations in pain sensitivity are a core clinical feature (i.e., allodynia, hyperalgesia), and epidemiological data show that the presence of these hyper- and hypoalgesic symptoms is strongly associated with the degree of pain reported by the subjects. There is also evidence that increased pain sensitivity may have an etiological role in at least some acute and chronic pain states.
Thus, though there is much work to be done before these relationships are fully understood, current evidence suggests that individual differences in pain sensitivity are of importance to the etiology, maintenance, and severity of clinical pain. It would therefore be unnatural to limit pain phenotypes to experimental pain measures. Rather, one would wish to include clinical measures that somehow capture this variation in pain sensitivity. As discussed in the succeeding text, lack of control over stimulus parameters (i.e., degree of pathology) is a serious limitation of clinical models, but experimental models have their own weaknesses in terms of validity for clinical pain. These approaches are therefore best viewed as complementary, and study designs are substantially strengthened by including phenotypes from both clinical and experimental domains.
- (3) Pain Scaling
To assess pain sensitivity, one must assess pain, which incredibly has not been done in many population-based studies of pain. This may in part be due to a common misunderstanding that subjective pain ratings cannot be relied to reflect the person’s actual pain experience: for instance, some subjects exaggerate their pain, or scales are interpreted and used differently by different subjects. There is considerable evidence that this skepticism is unwarranted. This includes validation through brain imaging, triangulation studies showing consistency in scale use across clinical and experimental pain, and high heritability of pain rating for certain types of pain. However, the most significant piece of evidence is that pain ratings of different experimental pain modalities are poorly correlated. This would not be the case if ratings were systematically biased, for instance, if some subjects tended to use scales more conservatively than others. However, for researchers using experimental pain measures, it is important to note that this argument is invalidated if different modalities are combined to an aggregate measure. Doing this would magnify whatever common variance exists across modalities, which may very well be a biased scale use.
Pain is most commonly scaled in the intensity domain, less frequently in the affective domain. There has been a rather long-winded debate about which instrument is best suited for this, with the 11-point Numeric Rating Scale (NRS) and Visual Analog Scale (VAS) as the chief contenders. For inexplicable reasons this debate never seems to settle, but there are good reasons to favor the VAS, including higher resolution and true ratio scale properties (i.e., that pain that is considered twice as strong by the subject is actually rated as twice as strong), neither of which is true for the NRS. Dichotomizing pain measurement will lead to a severe loss of power in GAS, as will do the use of pain scales with substantially lower resolution than the subject would be capable of producing given an adequate scale. The use of proprietary measures also limits comparability between studies, thwarting efforts to combine samples to achieve sufficient statistical power for genome-wide and next-generation sequencing studies. Thus, though almost any pain measurement is better than none, pain scaling should be done with a pain scale in common use, with adequate resolution and well-documented measurement properties, preferably the VAS. Aside from intensity and affective dimensions of pain, there are other aspects of interest. For chronic pain these include the duration and frequency of pain and the number of body sites in pain.
- (4) Heritability
The heritability of a phenotype can be defined as the proportion of the variance in the phenotype that is attributable to genetic variation in the population. For dichotomous phenotypes, such as diseases, heritability refers to the underlying risk of acquiring the phenotype and not to the overt phenotype itself. It should be noted that heritability may vary over populations, sex, age, and as a function of environmental exposure and is, strictly speaking, a characteristic of a phenotype as expressed in a given population, not of the phenotype per se.
Furthermore, as discussed in the succeeding text, heritability estimates can be strongly affected by measurement characteristics such as lack of reliability and stability over time or too broad or too narrow phenotype definition. In this sense heritability can be taken as an indicator of the quality of the phenotype and its suitability for GAS. So though this point shouldn’t be overstressed, phenotypes with moderate to high heritability are preferable where available.
- (5) Genotype–Phenotype Matching
Heritability is in itself an indicator, since high heritability cannot be achieved without a good genotype–phenotype match. Conventional factor analysis and related techniques also provide useful information about associations – albeit without parsing these associations into genetic and environmental components.
- (6) Reliability and Temporal Stability
It is therefore advisable that GAS employing experimental pain testing would include retesting of a subsample of a hundred or more subjects to document measurement characteristics of test as executed in the study.
The elderly may be less likely to form an optimal population, since many phenotypes tend to become less heritable in the upper age brackets as a consequence of accumulated environmental exposures.
- (7) Clinical Phenotypes
Unfortunately, a major issue with studying clinical phenotypes is that pain is confounded with disease severity. This may not be a major issue for candidate gene studies where assumption about gene function have been made a priori, but is a major concern for “hypothesis-free” approaches such as genome-wide association studies, aimed at discovering novel molecular mechanisms. Understanding the functional significance of novel findings will be vastly more difficult if it is not even known what physiological system they are associated with.
-
(8) Designing Clinical Pain Genetic Studies
-
(9) The Heritability of Specific Clinical Pain Conditions
For conditions where there was sufficient data to arrive at a reasonably firm conclusion, it was found that rheumatoid arthritis had a heritability of 52% in the United Kingdom and 65% in Finland (reanalysis of two large concordance studies), migraine had a heritability of 45% (meta-analysis), low back pain had a heritability of 34% (meta-analysis), and irritable bowel syndrome had a heritability of 22–27% (three large studies). Finally, several studies indicate that widespread pain/fibromyalgia has heritability of around 50%. Thus, there appears to be fairly large variation in the heritability of specific conditions. However, none of these studies actually measured pain severity beyond a simple yes/no question, making their relevance to pain genetics unclear.
- (10) Experimental Phenotypes
Unlike clinical phenotypes, experimental pain stimuli do not confound pathology with pain, since stimulus procedures can be standardized across subjects. Thus, experimental models provide a direct method of measuring pain sensitivity. Experimental methods are also the primary basis for several extended applications, such as brain imaging and pharmacogenetic studies. A final and huge advantage with these methods is that they have close parallels in animal models, paving the way to translational research of gene function. Though experimental methods are time consuming and therefore costly when applied in large samples, the advantages may well make it worth the effort.
However, there are important unresolved issues with experimental phenotypes as applied in GAS, which need to be resolved. First, there are a large number of experimental models, which vary across the intensity domain (pain thresholds, direct rating of suprathreshold stimuli and pain tolerance), modality domain (heat, pressure, cold, ischemic, etc.), temporal domain, and spatial domain. Human studies have shown that these pain models are poorly correlated with each other, and factor analysis points to stimulus modality as the organizing factor. Similar results have been shown with mice, where it has been demonstrated that the different pain modalities appear to be genetically distinct. In line with this, a human twin study found minimal genetic overlap between heat pain and the cold-pressor test. On the other hand, many chronic pain conditions show considerable comorbidity, suggesting common etiology. As mentioned earlier, nearly all the risk of having pain at different anatomical locations can be explained by one common heritable risk factor. So if many common types of clinical pain appear to have the same genetic basis, whereas the various experimental pain models have distinct genetic underpinning, then it is highly unlikely that all experimental models are equally suited for unraveling the genetic basis of clinical pain. The question that remains unanswered is therefore which experimental phenotype serves the best as a model for clinical pain.
The best way of approaching this question would be to conduct a large twin study including both experimental models and questionnaire data on clinical pain in order to estimate the genetic correlations between the experimental and clinical phenotypes. This would go a long way toward resolving the issue, at least as far as the relationships with common clinical phenotypes go. Until this is done, less conclusive evidence may be sought. This includes documenting the stability over time of experimental assays, directly comparing their phenotypic associations with clinical pain, and conducting prospective studies to approach an understanding of the causal direction between pain sensitivity and chronic pain. As mentioned earlier, the latter issue has been addressed by a few studies, but none of these have directly compared predictive value of different experimental pain models.
- (11) The Heritability of Experimental Phenotypes
Twin studies appear to show large differences in heritability between experimental pain models, ranging from 0% to 60%.
-
(12) Extended Phenotypes
-
(13) Practical Concerns
As genotyping becomes cheaper and the number of associations tested increases, the demands on sample size increase. Techniques that work well in small-scale clinical and laboratory studies may prove prohibitively time consuming and expensive in large samples. In addition to the financial and practical issues, safety becomes an important concern where thousands of subjects are included and must be given careful consideration.
- (14) Conclusions
Future GAS are likely to demand large samples with in-depth phenotyping of both clinical and experimental pain. Careful selection of phenotypes will be crucial for gaining significant and meaningful results from these studies. Among the core characteristics these phenotypes should have are moderate to high heritability and good stability over time. Clinical phenotypes will need to be defined in such a way that the confounding of pain with pathology is minimized. For experimental phenotypes, documentation of association with clinical pain, preferably from longitudinal studies, is important. More sophisticated measures of specific mechanisms or stages in pain processing are desirable but may need further development to be useful in GAS.
2.4 Genetic Contributions to Pain and Analgesia: Interactions with Sex and Stress
Most pain phenotypes are complex genetic traits that do not follow classical Mendelian patterns of inheritance. Two main phenomena characterize such traits – polygenic nature and interactions (gene X gene and gene X environment). Moreover, it is increasingly appreciated that genetic factors interact with other endogenous and exogenous variables to impact pain and analgesia. Regarding the former, sex differences in pain have been extensively documented, and genetic influences on pain can vary considerably based on the sex of the individual. Regarding the latter, psychosocial stress is known to affect pain responses, with higher levels of stress conferring increased risk of clinical pain. Importantly, gene X stress interactions have been shown to influence pain-related phenotypes.
- (1) Brief Overview of Sex and Gender Differences in Pain and Analgesia
Abundant epidemiological evidence reveals that the prevalence of many common chronic pain conditions is greater for females than males. Specifically, females show higher frequencies of most musculoskeletal pain conditions, migraine headache, and irritable bowel syndrome. Human experimental findings parallel these clinical results, with females consistently showing greater sensitivity than men to experimentally induced pain.
- (2) Brief Overview of Stress and Pain/Analgesia
stress-induced analgesia (SIA) and stress-induced hyperalgesia (SIH)
- (3) Sex X Gene Interactions in Pain and Analgesia
-
(4) Sex Chromosome-Linked Genes
- (5) Human Findings
For example, a large twin study indicated that the heritability of neck pain was higher for females than males. Candidate gene studies also suggest sex-dependent genetic associations. For example, COMT single-nucleotide polymorphisms (SNPs) and haplotypes were associated with pain scores in females but not males with major depression. In contrast, an SNP of the actin-binding LIM protein 3 gene (ABLIM3) was recently associated with cold-pressor pain in males but not females. Regarding analgesic responses, a recent study found that an SNP of the serotonin 2A receptor gene 5-HTR2A was associated with analgesic requirements following abdominal surgery in women but not in men.
We previously demonstrated that A118G SNP of the mu opioid receptor gene (OPRM1) was associated with pressure pain sensitivity, marginally more strongly in males than females.
- (6)Translational Findings
The Melanocortin-1 Receptor Gene (MC1R)
The Arginine Vasopressin Receptor 1A Gene (AVPR1A)
- (7) Summary
The interactions of sex, genotype, and stress represent a great challenge to any simple conclusions regarding the influence of any one of them on pain biology. For better or for worse, it is interactions that produce biology, and pain biology can only be appreciated in terms of such interactions. We “control for” them and ignore them at our peril.
3.Title: “Human Genetics of Pain (2019).”
James J. Cox, Ingo Kurth, and C. Geoffrey Woods. The Oxford Handbook of the Neurobiology of Pain
3.1 Abstract and Keywords
In the postgenomic era, single-gene mutations for numerous human Mendelian pain disorders have been described owing to advances in sequencing technology and improvements in pain phenotyping. The study of painless and painful inherited monogenic disorders has led to the identification of key genes that are needed for the normal development or function of nociceptive neurons. Genes that are covered include ATL1, ATL3, DNMT1, DST, ELP1, FLVCR1, KIF1A, NGF, NTRK1, PRDM12, RETREG1, SCN9A, SCN10A, SCN11A, SPTLC1, SPTLC2, TRPA1, WNK1, and ZFHX2. The study of some Mendelian disorders of pain sensing has the potential to lead to new classes of analgesic drugs.
Keywords: pain insensitivity, hereditary sensory neuropathy, hereditary autonomic neuropathy, chronic pain, voltage-gated sodium channel, Mendelian genetics, inherited pain disorders, monogenic
3.2 Introduction and Scope
Mendelian disorders of pain: human extreme phenotypes caused by one or two mutations, typically single-nucleotide changes, in a single gene. And, these changes (pathogenic mutations) cause a pain phenotype irrespective of other genetic background or mutations in other genes; that is, they are fully penetrant.
Transcriptional regulators PRDM12, ZFHX2, and DNMT1 localize to the nucleus within the dorsal root ganglion (DRG) soma.
RETREG1 (reticulophagy regulator 1) is found within the endoplasmic reticulum (ER) and is also a structural protein of the cis-Golgi body.
The serine palmitoyltransferase enzymes SPT1 and SPT2 are involved in sphingolipid biosynthesis and localize to the ER.
Atlastin proteins ATL1 and ATL3 regulate ER architecture. Voltage- gated sodium (Na ) channels are expressed at both the peripheral and central terminals and along axons of primary afferents.
WNK1 (WNK lysine deficient protein kinase 1) is a regulator of ion channels and also interacts with KIF1A (kinesin family member 1A), a kinesin family motor protein involved in axonal transport of synaptic vesicles.
DST (dystonin) is involved in intracellular transport and maintains cytoskeletal integrity.
IKAP (elongator complex protein 1) mutations are associated with neuronal migration defects and impaired neurotrophic retrograde transport.
Nerve growth factor (NGF) and its receptor, TRKA, are important for neuronal development and survival.
DHN = dorsal horn neuron.
Differences between humans and other species can be exposed, such as biallelic NTRK1 (neurotrophic tyrosine receptor kinase 1) or SCN9A mutations, which cause painlessness in humans but early lethality in mice.
3.3 Mendelian Causes of Painlessness
Three Mendelian human pain insensitivity disorders are reported whereby patients present with a normal intraepidermal nerve fiber density (i.e., nociceptors are present but are nonfunctional). These disorders, congenital insensitivity to pain caused by mutations in SCN9A or SCN11A, and Marsili syndrome caused by a mutation in ZFHX2 (zinc finger homeobox 2).
- (1) Na 1.7 Congenital Insensitivity to Pain
In 2006, remarkable pain-insensitive patients originating from northern Pakistan were described, with the causative gene, SCN9A, identified. Since then, significant efforts have been made by the pharmaceutical industry to target the encoded Na 1.7 voltage-gated sodium channel, with the aim of generating new potent analgesics.
- 1) Phenotype
Individuals with Na 1.7 congenital pain insensitivity have a complete inability to experience pain and are also anosmic (lack a sense of smell). With the exception of pain sensation, somatosensory functions are normal, with touch, warm and cold temperatures, proprioception, tickle, and pressure all correctly perceived.
- 2) Genotype
Na 1.7 congenital insensitivity to pain is an autosomal recessive disorder and was first reported to map to a region on chromosome 2 by autozygosity mapping in three consanguineous families from northern Pakistan. Candidate gene analysis identified SCN9A as a lead target, with previous mouse Scn9a conditional knockout studies in sensory neurons showing a significant pain-insensitive phenotype.
- 3) Pathogenesis
Studies of Na 1.7-positive neurons in the mouse hypothalamus have shown that a persistent sodium current mediated by Na 1.7 is critical for synaptic integration. Na 1.7 is predominantly expressed within dorsal root ganglia (DRG), trigeminal ganglia, sympathetic neurons, and olfactory epithelia.
Studies in conditional mouse Scn9a knockouts have helped to explain why null mutations cause anosmia and pain insensitivity. Using olfactory sensory neuron (OSN) conditional Scn9a knockouts, Weiss et al. showed that the OSNs were still electrically active and could generate odor-evoked action potentials but failed to initiate synaptic signaling to the projection neurons in the olfactory bulb.
Further insights into the consequences of a loss of Na 1.7 have been revealed using transcriptomic analyses of DRG from Scn9a knockout mice, which showed a significant upregulation of the endogenous opioid preproenkephalin (Penk). These links between Na 1.7 loss of function and opioid signaling may be used therapeutically as Na 1.7 blockers combined with low-dose opioids or enkephalinase inhibitors are reported to produce profound analgesia.
4.Title: “Understanding the Psychological, Physiological, and Genetic Factors Affecting Precision Pain Medicine: A Narrative Review (2021).”
4.1 Introduction
The overarching definition of precision pain medicine is that diagnosis and treat-ment can be customized to an individual’s specific risk profile. The goal of precision medicine is to maximize the accuracy by which patients are treated with existing treatment regimens and is informed through an understanding of the interrelation of an individual’s profile of characteristics, including genetics, environment, and lifestyle, with specific inclusion of phenotypes and biological markers.
4.2 Physiological Factors
QST is a collection of methods designed to measure patient response to various stimulation (eg, mechanical, thermal, cold, pressure) in order to evaluate somatosensory function as well as identify the nature/presence of hyperalgesia and allodynia. It is most commonly utilized to evaluate neuropathic pain conditions. The protocol with the best validation is the German Research Network on Neuropathic Pain (DFNS) battery, which can help determine detection and pain thresholds to both mechanical and thermal stimulation in addition to assessing for wind up pain. Following completion of this protocol, patients can be assigned a profile. Previous research in this area has shown that QST profiles can have tremendous overlap between different neuropathic conditions, indicating that the profile is not pain syndrome-specific but patient-specific, thus hopefully allowing for individualized treatment regimens.
4.3 Genetic Factors
Individual differences in the DNA sequence (genetics) and the structure of the genome (epigenetics) are estimated to account for up to 70% of the individual differences in pain sensitivity and susceptibility to chronic pain conditions in addition to affecting the response to pain-relieving treatments (eg, pharmacogenetics).
Single nucleotide polymorphisms (SNPs) are the most common variants and represent differences in the nucleic acid sequence at a given genomic location (ie, alleles) between individuals. The major allele is present in most of the population, and the less common (ie, minor) allele frequency varies but occurs in greater than 1% of the population. This type of genetic variation occurs approximately every 1000th nucleotide, so it is estimated that there are roughly 4–5 million SNPs in the human genome contributing to the significant phenotypic variation across the population.
There are several genes where SNP genotype has been associated with differences in pain severity or risk for development of a chronic pain condition. The relationship between pain susceptibility and variations within these genes may help to identify patients who are at risk for disproportionately severe pain or who are most likely to develop chronic pain. These relationships could not only help to identify those patients at the highest risk for pain but also point to specific targets for novel precision pain therapeutic development designed to address the underlying mechanism of risk. Arguably, the most well-defined example in this category of “pain genes” is COMT, encoding the enzyme catechol-o-methyltransferase (COMT) responsible for the breakdown of catecholamines. COMT SNP genotype is associated with the altered sensitivity to painful stimuli as well as the development of chronic pain conditions (eg, fibromyalgia, chronic widespread pain, irritable bowel syndrome, migraine headache) and may contribute to individual differences in morphine analgesic efficacy. COMT genotype is not currently being used to inform precision pain management in the clinical setting but, moving forward, it may help to identify patients at the highest risk of developing chronic pain after interventions like surgery and chemotherapy. Members of the family of voltage gated sodium channels responsible for action potential generation and propagation within pain-sensitive neurons include SCN8A-SCN11A, encoding Nav1.6, Nav1.7, Nav1.8, and Nav1.9 respectively. Variations within these genes were first implicated in monogenic disorders of altered pain sensitivity (eg, congenital insensitivity to pain, familial episodic pain syndromes, inherited erythromelalgia, and paroxysmal extreme pain disorder), but more recently associations have been identified for pain sensitivity in non-pathologic individuals and risk of developing chronic pain conditions. While genotyping for this group of genes is not currently being used clinically outside of diagnosis for monogenic disorders, the genetic and functional validation of these channels in human pain has led to the development of selective sodium channel inhibitors to replace traditional local anesthetics. In the future, the selection of sodium channel selective molecules could be tailored to the procedure as well as the patient’s genotype to improve pain outcomes.
4.4 Limitations of the Field
In fact, given that the impact of psychological, physiological, and genetic factors could have differential impact on chronic pain risk based on race, ethnicity, sex, and other socioeconomic factors, the application of new findings to diverse groups must be based on evidence-based medicine and not on the assumption that all groups will benefit equally from precision pain management strategies that work for others.
4.5 Moving Forward
Chronic pain continues to be a growing public health problem, requiring significant financial and health-care resources annually while negatively impacting the wellness and quality of life of millions. Identification of patient risk profiles by incorporating genetic and phenotypic data is key to the development of precision pain and analgesic medicine strategies. The evidence is clear that pain is an individualized experience and personalized and/or precision treatments could improve pain outcomes. As of this moment, however, we are still in the discovery phase with the goal of moving into evidence-based practice in the coming years. Once we have a clear understanding of the mechanisms that drive pain, we can then progress beyond the basic diagnosis and treatment of symptoms to the management of the underlying pathophysiology.
5.Title: “Genetic basis of pain variability: recent advances (2012).”
5.1 Abstract
An estimated 15–50% of the population experiences pain at any given time, at great personal and societal cost. Pain is the most common reason patients seek medical attention, and there is a high degree of individual variability in reporting the incidence and severity of symptoms. Research suggests that pain sensitivity and risk for chronic pain are complex heritable traits of polygenic origin. Animal studies and candidate gene testing in humans have provided some progress in understanding the heritability of pain, but the application of the genome-wide association methodology offers a new tool for further elucidating the genetic contributions to normal pain responding and pain in clinical populations. Although the determination of the genetics of pain is still in its infancy, it is clear that a number of genes play a critical role in determining pain sensitivity or susceptibility to chronic pain.
5.2 Introduction
Pain—the “unpleasant sensory and emotional experience associated with actual or potential tissue damage…” (International Association for the Study of Pain taxonomy)— is a necessary and informative sensory experience which encourages avoidance of danger and recuperative behaviours that promote healing and protection of an injured or diseased area of the body. In pathological conditions, the pain may no longer be useful and may produce more harm than good. Estimates suggest that 15–50% of the population is experiencing pain at any given time. Because pain is the most commonly reported symptom in clinical settings,4 5 individual differences in pain reporting could contribute to delayed or ineffective treatment of underlying disease in those with low sensitivity,8 while hypersensitivity could increase an individual’s reporting of pain leading to significant inordinate healthcare utilisation and unnecessary personal suffering, and may increase risk for a number of chronic pain conditions.
Though pain is often a normal part of the human condition (with rare exceptions), there is a high degree of inter-individual difference in pain responses and in reporting of pain in the clinical setting. This variability is likely due to the complex interaction of environmental and innate factors. For example, socioeconomic status and prior history of trauma/stress exposure have both been shown to modulate pain reporting and responding. Heritability estimates based on inbred strains of laboratory mice studies suggest that up to 30–76% of the variance in pain responding is explained by genetic factors. Race and ethnicity have also been shown to explain differences in reports of pain, with African American and non-Caucasian Hispanics typically reporting more pain than Caucasians within the same clinical populations. In addition, gender has been shown to affect pain thresholds as well as pain reporting in a clinical setting, with women typically reporting greater pain than men. Even when the variability explained by each of these factors is accounted for, individual differences remain and genetic variation has been shown to explain a significant portion of this remaining variability. While understanding the phenomenon has basic intellectual value, the influence of genetic variation on pain variability is also highly clinically relevant as it may lead to more individualised care for patients and the identification of novel therapeutic targets.
Early research using twin and family studies has established the heritable nature of both experimental and clinical pain. These studies were instrumental in identifying familial trends for pain conditions and rare disorders in which pain responding is significantly altered. Linkage and association studies have pinpointed a number of genes responsible for heritable conditions involving alterations in pain. The hereditary sensory and autonomic neuropathies (HSAN I–V) are a family of such syndromes in which pain perception and responses are significantly reduced or absent due to mutations in single genes.
Individuals diagnosed with HSAN often exhibit progressive injuries to the areas of the body affected (ie, ulcerations, joint deformities, etc) but do not report discomfort as a result. Other Mendelian heritable conditions are associated with increased pain including erythermalgia, familial hemiplegic migraine, and paroxysmal extreme pain. While these types of pathological conditions add to our overall knowledge regarding pain processing, they do not necessarily give insight into variations within the general population. There is growing evidence that in order to understand the genetics of pain, pain must be considered a complex phenotype or trait resulting from complex polygenic and environmental contributions. Now, more than ever, researchers are focusing on the genetic contribution to normal variation in pain reporting and responding, as this may facilitate translation of basic science findings into pain treatment protocols individually tailored to a patient’s pain risk or resilience.
Research into the genetics of pain in humans utilises a number of methodologies to identify genetic correlates of behaviour. Identifying mutations may explain rarer inherited pain syndromes, but the application of these findings to variations in the general population has been less fruitful. Twin studies offer an opportunity to evaluate polygenic inheritance. Twin studies and other studies suggest that 30–60% of the variation in chronic pain syndromes may be due to heritable factors. Recently developed genome-wide arrays allow for the objective unbiased evaluation of the association of human pain phenotypes with single nucleotide polymorphisms (SNPs) across the entire genome, including variations in the number of copies of a gene that an individual has (copy number variation (CNV)).
4.3 Genetic correlates of pain: recent progress
Significant individual variability is observed in both pain threshold and in susceptibility to chronic pain conditions, and a portion of this variation can be explained by variation within specific genes. Single functional SNPs or combinations of SNP alleles that tend to be inherited together (haplotypes) can contribute to increased or decreased susceptibility to pain. One of the most extensively studied pain candidate genes is catechol-O-methyltransferase (COMT), which is known to be involved in the inactivation of dopamine, epinephrine, and norepinephrine neurotransmission and associated with variations in experimental and clinical pain behaviour. Four SNPs have been identified that may contribute to a haplotype characterised by differences in COMT metabolic enzyme activity that is inversely correlated with alterations in pain perception. Additionally, a single protective haplotype has been related to increased enzymatic activity, decreased pain sensitivity, and reduced risk for temporomandibular joint disorder, a common musculoskeletal pain syndrome. While genomic variation in COMT affects RNA stability and protein translation and affects pain through variations in neurotransmitter metabolism, SNPs in the μ-opioid receptor gene (OPRM1) alter pain sensitivity through variations in receptor function. Relationships between OPRM1 and pain sensitivity and efficacy of opioid analgesics have been shown in a number of experimental and clinical populations. Furthermore, recent data show that genetic mutations in COMT and OPRM1 may interact and act synergistically to affect morphine analgesia and side effect burden. Additional evidence comes from the identification of SNPs within the melanocortin 1 receptor (MC1R) gene that are associated with pain sensitivity and μ-opioid analgesia in certain populations.
- (1)Genes affecting neurotransmitter systems may determine pain phenotypes
Mechanisms of variability related to neurotransmitter systems are of particular clinical relevance due to the number of pharmacological agents already designed to act on them (ie, agonists and antagonists used for other purposes).
Polymorphisms within GCH1, the gene encoding GTP cyclohydrolase 1, have been shown to form a protective haplotype with reduced sensitivity to both clinical and experimental pain measures. GTP cyclohydrolase 1 is the rate limiting enzyme in tetrahydrobiopterin (BH4) formation, a necessary step in the biosynthesis of serotonin, dopamine, norepinephrine, epinephrine, and nitric oxide, all of which have been shown to have a significant role in pain processing. As with COMT, it is unclear which of the target neurotransmitters is critically affected and by what mechanism the genetic differences result in an alteration in pain phenotype. However, it is important to note that it is not clear whether GCH1 plays a role in modulating all pain phenotypes. However, other genetic loci that contribute to catecholamine synthesis and transmission have been implicated in a number of pain phenotypes. Most recently, an association has been revealed between polymorphisms of the serotonin transporter gene (SLC6A4) and experimental thermal pain thresholds76 as well as the conditioned and emotional82 modulation of experimental pain.
Recent data suggest an association between genes encoding for two neurotransmitter receptors, one for serotonin and one for epinephrine, and an increased risk for developing CWP in which pain is the primary symptom of concern. Taken together with previous data, these findings suggest that genotype differences can result in alterations in neurotransmission, which can in turn contribute to variations in pain reports within normal and clinical populations.
- (2)Genetic determinants of ion channel function contribute to pain susceptibility
While alterations in neurotransmission directly affect the messages sent and received by neurons, alterations in ion channels can alter the transmission of messages received by augmenting or decreasing neuronal excitability. Sodium, potassium, and calcium channels are known to play a vital role in initiation and propagation of intracellular signals in neurons, including primary nociceptors that innervate peripheral tissues and are activated by noxious stimulation to propagate nerve impulses towards the spinal cord.
Recent findings suggest an SNP within the SCN9A gene that encodes the α subunit of the voltage gated sodium channel NaV1.7 may play a role in determining risk for chronic pain conditions as well as variation in pain responding within normal populations. In a mixed cohort of sciatica, osteoarthritis, pancreatitis, lumbar discectomy, and phantom limb pain patients, increased pain was associated with the presence of an A allele at SNP rs6746030 within SCN9A (chromosome 2q24), resulting in an amino acid change from arginine to tryptophan at position 1150 of the Nav1.7 voltage gated sodium channel. This same SNP was associated with decreased thresholds for a composite measure of experimental pain (combined thermal, mechanical, ischaemic, and temporal summation of thermal stimuli). The subunit encoded by this gene is widely expressed in nociceptors and loss of function alleles have been implicated in congenital autosomal recessive channelopathies characterised by an inability to feel pain (channelopathy associated insensitivity to pain). Further implicating SCN9A in modulation of pain, primary erythermalgia and paroxysmal extreme pain disorder—both disorders characterised by an increase in pain sensitivity—are the result of autosomal dominant mutations shown to facilitate activation of NaV1.7.
Intracellular communication within the nervous system depends on the movement of sodium and potassium ions, and both populations of ion channels have recently been linked with alterations in pain sensitivity.
Two genes related to calcium channel function have been recently identified for their association with altered pain sensitivity. Individuals homozygous for two minor allele variants within CACNA2D3 (C/C at SNP 6777055 and A/A at SNP rs1851048), encoding for the alpha 2 delta 3 subunit of voltage dependent calcium channels, exhibit reduced sensitivity to acute noxious heat as well as with lower risk for chronic back pain following surgical intervention for discogenic disease.
- (3)Disease related genes play a role in pain responding
Susceptibility to a disease and the pain associated with that disease may have overlapping genetic contributions. On face value alone, it stands to reason that when variation in a gene is associated with an increase in disease severity, one might expect higher pain reporting. However, it is not always the case that disease severity and disease induced pain go hand-in-hand. In fact, there is often pronounced variability of pain reports even within a seemingly homogenous population. Moreover, previously identified ‘pain genes’ may affect pain without altering disease process, but this does not detract from their inherent importance for clinical practice. In short, though pain and disease may be somewhat related in nature, it is important to address each effectively, and genetics may offer a tool for maximising quality of life by decreasing pain as a separate focus during treatment.
4.4 Pain GWAS: progress and pitfalls
Our understanding of the genetics of human pain is rapidly growing and several recent GWAS have offered a glimpse of what is to come in terms of pinpointing distinct genetic contributions to risk and severity of pain syndromes.
Polymorphisms in transient receptor protein channels, such as those encoded by TRPM1, have been associated with neuropathic pain in a rodent model of peripheral nerve injury and may, therefore, show promise as candidate genes for pain susceptibility across models.
The genome-wide approach to studying human pain is still in its infancy due to the complexities involved with the potentially heterogeneous populations with a given diagnosis, the expense of genotyping samples from large cohorts, and the analysis of data that may not be suited for standard statistical analyses. Using this methodology, it may be possible to identify novel mediators of pain beyond those molecules discussed in the neurobiology literature and/or prioritise among the existing pain targets for further mechanistic studies and drug discovery with data collected specifically in human subjects. Clinically, GWAS could provide a tool for precise classification of pain syndromes based on shared genetic underpinnings, thereby enhancing both diagnosis but also treatment. Understanding the genetic contributions to patient risk for pain, chronic pain susceptibility, and/or the expected efficacy of analgesic therapy would allow for truly individualised patient care.
Significant relationships have been found between pain related traits, behaviours and candidate genes, but it is important to note that the literature available does not paint a picture of absolute certainty. While we have focused herein on significant relationships found in association studies, there are numerous studies in which associations between the same genetic targets and pain fail to replicate these significant findings.
A number of factors may be at work in these seemingly inconsistent findings. First, the methods used to analyse early genetic associations with complex traits likely resulted in the identification and reporting of spurious relationships.113 As the field has developed, bioinformatics techniques have evolved that reduce the risk of false positive reports. Aside from the methods used to collect and analyse the data for genetic associations, there are also some basic issues related to the populations used for these studies. There is substantial population variability between studies, defined by differences in demographics as well as differences in diagnoses and pain status. For example, it remains to be seen whether genetic associations that exist for one type of chronic pain (eg, chronic post-mastectomy pain) are also true for other types of chronic pain (eg, lower back pain, cancer pain, phantom limb pain). This may be, at least in part, due to the relatively small number of studies published using genetic association methods to assess human pain that are available for comparisons and hypothesis generation. Moreover, the lack of consistent replication across human studies may be due to inadequate power, population heterogeneity in a single study (ie, based on differential disease diagnosis, ethnicity, gender, etc) or differences in the method of measurement and reporting of pain across studies.
Importantly, one notable factor that has been somewhat overlooked is the potential for independent genetic associations with specific pain behaviours or pain states. Findings from animal studies would suggest that some specificity of genetic associations with modality or type of pain is expected and human studies have shown non-overlapping genetic associations with different pain modalities. As seen in figure 1, there is a lack of evidence for specificity of genetic associations with specific types of pain in humans. Experimental pain studies suggest that pain specific genetic associations are likely, but the translation of these findings to clinical pain has not yet been achieved. For instance, studies combining several cohorts (defined by diagnosis and/or pain outcome) may shed light on common mechanisms involved in multiple pain states, but may also fail to show significant genetic associations that are specific to only one of the cohorts in question. This circumstance could result in an artificial narrowing of the candidate gene list for subsequent hypothesis testing, and could lead to over-generalisation and false assumptions in future studies. The challenge at hand, therefore, is how to efficiently increase power in human pain studies to test specificity hypotheses in cohorts that represent different pain populations.
4.5 Translational potential of genetic association studies
The ultimate value in understanding the genetic determinants of pain is to be able to reduce suffering in human populations. While the flow of information from basic and clinical science studies is beginning to increase, there has not been a boon of genetic testing for use in risk assessment and diagnosis of pain in typical healthcare settings. There are, however, a number of genes that seem to have the most translational potential and may represent key tools in diagnosis and treatment of pain in the future. These can be roughly divided into three categories of translational application based on the association between the gene and pain phenotype: pain facilitating alleles, pain protective alleles, and alleles related to analgesia.
A number of recent associations suggest that certain polymorphisms act to facilitate or increase pain; the most recent genes of interest within this group include KCNS1, SCN9A, ADRB2, H2TRA, CACNG2, and IL16 (for details, see above). Genetic association studies indicate that these genes contribute to an increase in pain sensitivity and, in many cases, an increased risk for developing chronic pain conditions. As a result, the combined genotype for an individual at these multiple loci could give insight into risk for pain following treatment in the clinical setting. For example, if a patient was considering the benefits and costs of an elective surgical procedure, his or her genetic predisposition (or lack thereof) for developing neuropathic pain afterwards could be used as a factor in the decision.
Another family of alleles has been identified as conferring pain protection or a decrease in pain; the most recent candidates in this category include COMT, OPRM1, TRPV1, MC1R, GCH1, and CACNA2D3. These loci are associated with reductions in pain and/or resiliency to develop chronic pain. Thus, they could be of significant clinical importance to guide physicians and patients in determining who might be suitable for more aggressive treatment plans.
Finally, it is important to note that there are several alleles that have been shown to modulate the efficacy of analgesic agents in the treatment of pain. Polymorphisms in COMT, MC1R, and OPRM1 have been associated with resistance to the effects of analgesics. Polymorphisms within COMT61 and OPRM1 result in decreased morphine efficacy, while mutations of MC1R are associated with a reduction in the analgesic effects of lidocaine administration. Interestingly, other polymorphisms within OPRM1 are associated with decreased morphine side effect sensitivity—that is, decreased pupil constriction and decreased respiratory depression. Conversely, polymorphisms within two other genes, CYP2D6 (encoding for cytochrome P450 enzyme involved in metabolism of opioids) and ABCB1 (encoding for the P-glycoprotein transporter), have been associated with increased side effects following opioid administration. COMT haplotype has recently been associated with the efficacy of the β-adrenergic antagonist propranolol for pain reduction in patients with temporomandibular disorder. Interestingly, those with increased risk for chronic pain as a result of s-allele carrier status in the 5-HTTLPR of SLC6A4 exhibited better analgesic effects following opioid administration. While a risk assessment based on an individual’s pain related genotype is a long term translational goal, it may be more practical in the short term to identify those with a genotype suggestive of increased analgesic resistance and/or increased risk of negative side effects when designing pharmacological pain management plans, since these comprise much of the currently available arsenal used to fight pain.
6.Title: “Progress in genetic studies of pain and analgesia (2009).”
6.1 INTRODUCTION
Pain is a fundamental experience characterized by an unpleasant physical perception and corresponding emotional state. Pain is biologically adaptive in that it signals actual or potential tissue damage, which evokes withdrawal and/or recuperative behaviors.
6.2 INDIVIDUAL DIFFERENCES IN PAIN AND PAIN INHIBITION
One of the major reasons why pain relief remains such a challenge is the robust interindividual variability that exists in sensitivity to pain, the propensity to develop chronic pain conditions, and the response to analgesic manipulations. A surprising but potentially useful fact is that individual differences in laboratory pain sensitivity are predictive of clinical pain severity and response to treatment.
- (1)Heritability of Pain and Analgesia
Trait (phenotypic) variability is a result of variation in either the genetic (genotype) or environmental milieu, as well as the complex interaction of the two, which includes the increasingly appreciated role of epigenetic factors.
1)Twin studies
Unlike family history designs and ethnic comparisons, the twin study methodology (albeit with some caveats) is able to estimate heritability (h2) of a trait, that is, the proportion of trait variance due to inherited genetic factors. Any number of experimental and clinical pain traits have been so studied, with widely varying estimates of heritability ranging from essentially zero to 68%. The heritabilities of various modalities of experimental pain in the most modern studies are quite comparable to those obtained in systematic studies in mouse models. In many of the clinical studies, it is difficult to know whether what is being estimated is the heritability of developing the painful pathology, or the heritability of the pathology being painful. It is, of course, quite likely that different sets of genes underlie each.
6.3 UNCOVERING THE GENETIC DETERMINANTS OF PAIN AND ANALGESIA USING LABORATORY RODENTS
- (6)Limitations of Animal Models
However, major criticism of animal models of pain more generally stems from the promising targets identified in animals that failed to demonstrate clinical efficacy in human trials. It is unclear, however, whether the blame is properly placed on the animal research, on the trials themselves, or on the bad luck of unanticipated toxicity.
6.4 ASSESSING GENETIC FACTORS IN HUMAN PAIN AND ANALGESIC VARIABILITY
- (2)Genetic Association Studies of Pain and Analgesia in Humans
We are aware of at least 23 genes associated with experimental pain, clinical pain, or analgesia (excluding headache/migraine genes).
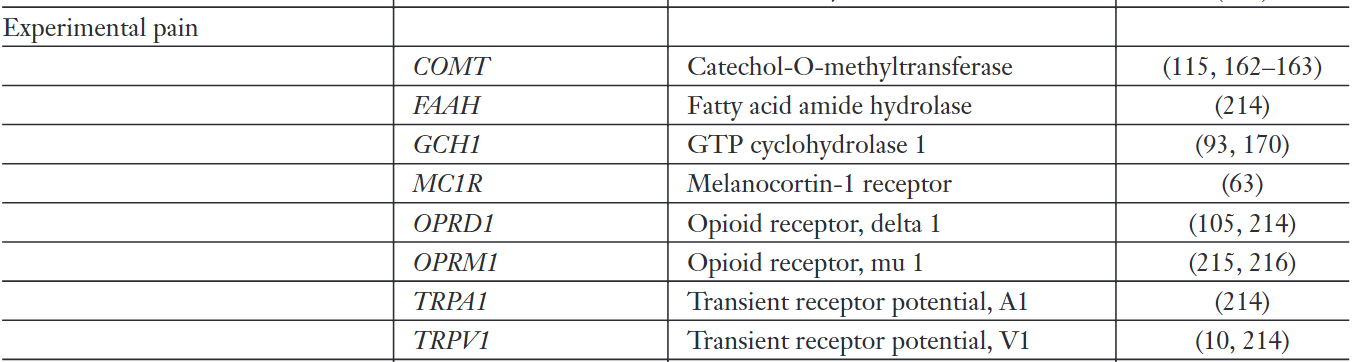
- (3)Do Genetic Association Studies Replicate?
The reporting of genetic associations with pain and analgesia has led to great excitement in the pain field, as it promises a revolution in our understanding of the risk factors for chronic pain development. This excitement is tempered by the reality of slow progress toward replication, contradictory data upon replication, and the modest percentage of the trait variance accounted for by the associated genes. Perhaps pain genetics is indeed extraordinarily heterogeneous with respect to study population and pain modality (as the animal research would predict), in which case we have much, much more work to do. Alternatively, the field of pain genetics may be riddled with false-positive and/or false-negative findings.
The current controversies in the human pain genetics field are not hugely surprising given the prior experiences of other fields in which many genetic association studies have been performed. In fact, this technique is widely known to be problematic. In a comprehensive review of over 600 reported genetic associations of common variants and disease, the authors found that of the 166 putative associations studied three or more times, only six were consistently replicated. Potential reasons for the lack of replication include population stratification, under- powering (the “winners curse”), too-broad (and thus ill-defined and possibly misattributed) phenotypes, and various types of bias. Solutions have been proposed for many of these problems, but have not yet been thoroughly implemented in the pain field.
- (4)Prospects for Genome-Wide Association Studies of Pain and Analgesia
The systematic nature of the GWAS approach might be particularly advantageous for human pain genetics, as a large number of high-priority candidate genes for neuropathic pain have been studied without success. In addition, GWAS studies are considerably more heuristic than the candidate gene studies currently dominating human pain genetics.
The rewards of such an effort aimed at pain and analgesia would likely be immense, and eventually lead to new treatments both for pain and true individualized medicine. In the best of all possible worlds, researchers would proceed by identifying relevant genes/proteins in humans (thereby proving their relevance), studying the roles played by these molecules in animal models, and then using this information to provide better treatments for those in pain.
7.Title: “Pain sensitivity risk factors for chronic TMD: descriptive data and empirically identified domains from the OPPERA case control study (2011).”
Different modalities of experimental pain stimulation (ie, me-chanical, thermal) recruit different groups of nociceptors and activate only partially overlapping CNS pathways. Furthermore, different pain assessment measures (ie, thresholds, suprathreshold ratings, TS) engage different aspectsof pain perception and thus, at some level, different CNS processes.
8.Title:”Genome-wide association reveals contribution of MRAS to painful temporomandibular disorder in males (2019)”
Abstract
Painful temporomandibular disorders (TMDs) are the leading cause of chronic orofacial pain, but its underlying molecularmechanisms remain obscure. Although many environmental factors have been associated with higher risk of developing painfulTMD, family and twin studies support a heritable genetic component as well. We performed a genome-wide association studyassuming an additive genetic model of TMD in a discovery cohort of 999 cases and 2031 TMD-free controls from the Orofacial Pain:Prospective Evaluation and Risk Assessment (OPPERA) study. Using logistic models adjusted for sex, age, enrollment site, andrace, we identified 3 distinct loci that were significant in combined or sex-segregated analyses. A single-nucleotide polymorphism onchromosome 3 (rs13078961) was significantly associated with TMD in males only (odds ratio52.9, 95% confidence interval: 2.02-4.27,P52.231028). This association was nominally replicated in a meta-analysis of 7 independent orofacial pain cohorts including160,194 participants (odds ratio51.16, 95% confidence interval: 1.0-1.35,P52.331022). Functional analysis in human dorsalroot ganglia and blood indicated this variant is an expression quantitative trait locus, with the minor allele associated with decreasedexpression of the nearby muscle RAS oncogene homolog (MRAS) gene (beta520.51,P52.4331025). Male mice, but not femalemice, with a null mutation ofMrasdisplayed persistent mechanical allodynia in a model of inflammatory pain. Genetic and behavioralevidence support a novel mechanism by which genetically determinedMRASexpression moderates the resiliency to chronic pain.This effect is male-specific and may contribute to the lower rates of painful TMD in men.
We found that TMD was associated with single-nucleotide polymorphisms (SNPs) in 7 genes:COMT, an enzymecatabolizing catecholamines;HTR2A, one of the receptors forserotonin;NR3C1, the glucocorticoid receptor;CAMK4, thecalcium/calmodulin-dependent protein kinase;CHRM2, themuscarinic acetylcholine receptor;IFRD1, the interferon-related developmental regulator; andGRK5, the G-protein-coupledreceptor kinase.
The aims of this study were to undertake the first discoveryGWAS of examiner-verified chronic TMD, evaluate generalizabilityofthe findingsthroughindependent replication,and investigate thefunctional biology of genetic markers that emerged from discoveryand replication phases.
9.Title: “Genetic components of human pain sensitivity: a protocol for a genome-wide association study of experimental pain in healthy volunteers (2019).”
Abstract
Introduction Pain constitutes a major component of the global burden of diseases. Recent studies suggest a strong genetic contribution to pain susceptibility and severity. Whereas most of the available evidence relies on candidate gene association or linkage studies, research on the genetic basis of pain sensitivity using genome-wide association studies (GWAS) is still in its infancy. This protocol describes a proposed GWAS on genetic contributions to baseline pain sensitivity and nociceptive sensitisation in a sample of unrelated healthy individuals of mixed Latin American ancestry.
Methods and analysis
A GWAS on genetic contributions to pain sensitivity in the naïve state and following nociceptive sensitisation will be conducted in unrelated healthy individuals of mixed ancestry. Mechanical and thermal pain sensitivity will be evaluated with a battery of quantitative sensory tests evaluating pain thresholds. In addition, variation in mechanical and thermal sensitisation following topical application of mustard oil to the skin will be evaluated.
10.Title:”Effect of Human Genetic Variability on GeneExpression in Dorsal Root Ganglia and Associationwith Pain Phenotypes (2017).”
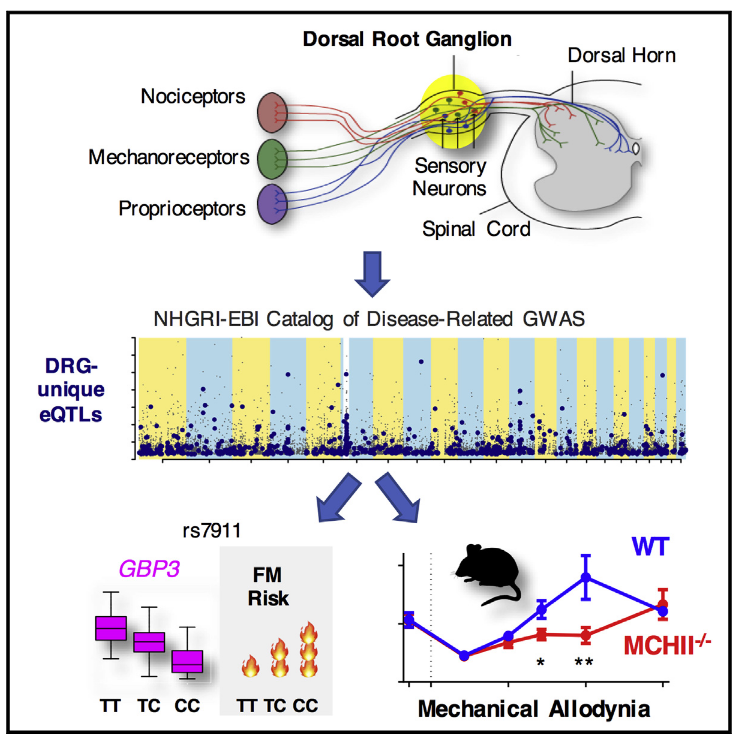
Pain is the primary reason for seeking medical assistance, but its underlying causes remain difficult to identify and remediate. Approximately half of the inter-individual variability in the experience of pain, risk of development of chronic pain, and responses to treatment are attributable to genetic factors. Identifying these genetic elements will improve diagnosis and facilitate the development of personalized therapies for people afflicted with chronic pain.
Genome-wide association studies (GWAS) have successfully identified genetic variants affecting risk for a wide range of human psychiatric and neurological conditions. The assessment of human chronic pain conditions using GWAS is an emerging but rapidly growing field. The major difficulty in the interpretation of GWAS results derives from the fact that most genetic variants, a majority of which are single nucleotide polymorphisms (SNPs), are not protein-coding, thus seldom provide a simple, straightforward molecular-level explanation. Other than producing coding changes, alternative alleles can affect gene function through their effecton mRNA expression level or splicing. SNPs associated with the expression level of a gene or an exon are called expression quantitative trait loci (eQTLs). Both SNPs that produce local expression effects (cis-eQTLs) and SNPs that affect expression levels of distant genes (trans-eQTLs) have been found to be functional. More than half of the known eQTLs are tissue-specific, making the identification of eQTLs in tissues relevant to disease pathogenesis a crucial matter.
The perception of pain and other somatosensory experience result from the relay of neural signals from the periphery into the CNS. Primary afferent neurons, which have their cell bodies located in dorsal root ganglia (DRG), convey sensory information (pain, itch, touch, pressure, vibration, and temperature). DRG neurons are heterogeneous and can be classified according to their size, neurochemical markers, stimulus modality, and gene expression profiles. DRG are situated at the junction between the spinal nerve and the dorsal root. They are maintained by a network of permeable capillaries lined by endothelial cells and are held together by a connective tissue capsule. In addition, resident T-lymphocytes and macrophages are found in DRG, increasing in number following injury. The perception of acute pain results from the activation of specialized DRG neurons referred to as ‘‘nociceptors’’. Increased nociceptor excitability following nerve injury and/or exposure to inflammatory mediators contributes to the hyperalgesia and allodynia often observed in chronic pain states. Due to their critical role in initiating, detecting, and perpetuating pain and related sensations, DRGs are a primary tissue target for analgesics, anesthetics, antipruritics, and neuro-stimulation procedures.